Questions mock defense
1/32
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
33 Terms
Of all compounds used in your thesis, which one is the most dangerous and why?
Many compounds required special caution, such as LDA because its strongly basic, moisture-sensitive and very toxic.
Also sodium azide is fatal if swallowed and releases toxic hydrazoic acid in contact with acid
hydroxylamine-O-sulfonic acid causes acute toxicity
But if I had to pick one, I would say bromine because it combines volatility (the bottle didn’t have a septum), severe corrosivity and is fatal when inhaled.
Also all michael acceptors used are toxic as well
You briefly mention toxicity related to host-directed antivirals and later on you mention that many kinases are dispensable for host survival, while their inhibition may disrupt multiple stages of the viral life cycle. Could you expand on the dangers of this strategy and the risk of targeting kinases?
The danger is that we are not targeting the virus but the host itself. Kinases are key regulators in cellular pathways, so inhibiting them not only prevents viral replication and spreading but also disrupts cellular processes. So inhibiting can cause harm to the body.
Therefore this inhibition is meant to be temporary as these compounds are metabolized and excreted by the body. This temporarily prevents viral replication and spreading and therefore gives the body’s immune system enough time to regain control; the challenge is therefor to find an effective but safe therapeutic window.
for small molecule inhibitors this can range from a few hours to a day and some more active metabolites even several days.
You mention a chalcogen bond being present in the crystal structure of 4Y8D. How can you recognize a chalcogen bond
This is a directional non-covalent interaction between chalcogen atoms (O, S, Se, Te) and electron rich atoms (lewis bases)
chalcogen atoms can have a region of positive electrostatic potential along the extension of the covalent bond called a sigma hole with which electron rich atoms can interact
in our case it is between the sulfur of the isothiazole and the oxygen of a backbone carbonyl
you can recognise them since their distance is usually smaller than the sum of their van der waals radii and the donor lies along the extension of the chalcogen covalent bond
in our virtual screening the chalcogen interaction was not included since common docking tools such as autodock fail to represent sigma holes.
Can you suggest a plausible formation of the formation of a carboxylic acid
so for the nitrile: (the nitrile and the aromatized starting material with aldehyde intact have similar molecular weight 189 and 190)
the amine is more prone to react at the aldehyde forming an oxime which after H-abstraction and elimination of the O leaving group yields the nitrile
Formation of the carboxylic acid is special
possibly it forms out of the nitrile itself through nitrile hydrolysis
first a ritter reaction towards the amide
followed by replacemtn of teh amine by hydroxyl group
Or some kind of reaction could occur where the hydroxylamine acts an oxidans like a hydride shift.
Aromatization
the benzylic hydrogens are readily removed due to conjugation with the aromatic system, the aromitzation is thermodynamically favorable, so under prolonged heating and perhaps the presence of oxygen and possilbe hydroxal amine derived oxidizing or radical species, this could occur.
Azide approach still suffer from low yields, how would you increase the speed of the addition elimination reaction or ringclosure
for the addition elimination reaction, the addition is the rate limiting step, therefor using lewis acids could help increase reactivity
For the ringclosure, UV light is known to convert azides into nitrenes which could therefore accelerate the process because this is the reactive synthon
How can you see the difference between the E and Z isomers
they showed similar peaks but slightly shifted where the E-isomer was a little lower in chemical shift and had much lower intensity
however only the aldehyde and vinyl protons were distinguishable because the aromatic peaks overlapped
also these products were already reported in literature so I could easily compare the chemical shifts, for the unsubstituted acetophenone, also the E-isomer peaks were reported
One interesting thing though, was that the acetophenone showed dd’s for both the vinyl and aldehyde protons which was not reported
likely due to long range coupling with an aromatic proton (no W coupling one carbon to much and not the right orientation)
Your compounds are meant to be used as inhibitors can you briefly evaluate the compound you have drawn in figure 16 by their durg-likeness.
they abide lipinski’s rule of 5
less than 5 H-bond donors
less than 10H acceptors
Mw lower than 500 g/mol
lipophilicity was not tested
although many contain an alcohol which helps with solubility
Fsp3 values of the final compounds were between 0.4 and 0.5 exceeding the commonly cited drug likeness treshold of 0.42 a critereon reportedly met by 84% of marketed drugs
although this was mainly due to sp3 carbons in the substituents but were delibirately chosen for this purpose
the tetralones only contain 2 sp3 carbons which are still restrained by the overall ring fusion, but the whole planarity of the molecule is gone and more flexible and therefore more likely to accommodate the geometry of the binding pocket
Mechanism of the Ullman coupling and explanation of the dimer formation
deanol is deprotonated by the phosphate base and coordinates with Cu(I) species already present form the CuI salt or generated in situ from metalic copper
the aromatic bromide oxidatively adds to the Cu(I) deanol complex
this creates a 4 coordinated Cu(III) species
than the bromine ligand is exchanged for the amine of L-prolinol
L-prolinol is made from chemically reducing L-proline which is a naturally occuring amino acid found in plant sources and animal products
than reductive elmination yields the aromatic amine coupled product and regenerates the Cu(I) deanol species
reductive elmination with the deanol ligand could cause side reaction (but were not observed)
Dimerization
Another aryl copperspecies in the reaction mixture can excahnge its coordinated aryl moeity to antohter aryl coordinated copper species through transmetallation, no two aryl moieties aer coordinated to the same Cu(III) complex and reductive elimination can occur.
Another patway is that after oxidative additon, reductive elimination occurs between other coordinated species leading to an aryl coordianted Cu(I) species, now oxidative addition can again occur to this species leading which also generates the diaryl Cu(III) species
Ulmann coupling mechanism have also not only been proposed through this Pd like mechanism but also through radical type pathways that somehow two aryl radical species formed may combine.
Why Metallic Cu?
The paper used a mixture of both Cu and CuI, they reported a similar ulman coupling using 2-halothiophenes where they originally worked on 2-iodothiophenes where they only used metallic copper, so the Cu(I) species was generated in situ which gave better yields with fewer byproducts. (also all metalic copper was consumed after the reaction, no copper precipitate proving the Cu(I) is formed.
the bromothiophenes however are less reactive so only using Cu gave an induction period, meaning the formation of the active copper species was slow. Therefor CuI is added to provide Cu(I) directly. they also tested the reaction without metallic copper and only CuI but this gave lower yields proving that the metallic copper still has some catalytic effect.
Why do you think DBU made the difference
Base | Conjugate acid | pKₐ (approx.) |
|---|---|---|
NaOtBu (t-BuO⁻) | t-BuOH | 18 |
DBU | DBUH⁺ | 12–13 |
K₂CO₃ (CO₃²⁻) | HCO₃⁻ | 10.3 |
K2CO3 is a weaker base and had poor solubility in DMF providing heterogenous conditions
it was probably not able to deprotonated the amine and therefore only the addition elimination product was observed
NaOtBu provided still heterogenous conditions but was a much stronger base, and although SM was completely consumed no product was observed
DBU had a basicity in between the two
it is non nucleophilic
soluble in organic solvents allowing the reaciton to occur under homogenous conditons, which may have promoted the reaction because of more efficient interactions with the substrate
How does the trimerization occur and did you observe it?
It occurs because the C=S bond is strongly polarized so a thioaldehyde molecule can act as an electrophile at the carbon and as nucleophile at sulfur, this gives a head to tail type self addition and after three units combine a stible trimer is formed
I did not observe them myself because the procedure was already optimized to 70 °C to convert the trimers back to monomers
These can complicate workup as these trimers can decompose on column to the thioaldehyde monomers, potentially contaminating all fractions
You suggest O-tosylhydroxylamine as a possible N-source, how would this react with your molecules if you consider reaction in scheme 24? Would this react differently than the other N-sources employed
yes as depicted in the scheme, there’s a high chance that the amine will form an imine with the aldehyde which is probably also the main problem with the other hydroxylamines, but this process is reversible, so potentially running longer and under thermodynamic conditons could yield the additon elimination product
it has also been pointed out to me recently that the this compound is very reactive and unstable at room temperature and prone to explosive decomposition, mainly because it’s activated because the tosylate is a good leaving group
so with our goal to replace the unstable explosive azide, on hindsite this probably not the best solution
how was the biological data measured
The compounds were tested in a dose-response ADP-Glo Kinase assay.
the compounds are first incubated with the kinase with different concentrations (in DMSO) of the compounds to allow binding with the kinase
ATP and a substrate that gets phosphoryated were added to start the phosphorylation reaction
non inhibited kinases convert ATP to ADP (the stronger the inhibitor the less ADP produced)
After incubation, ADP-Glo is added which steps the reaction and removes remaining ATP
Then a kinase detection reagent is added that turns the remaining ADP into a luminescence signal
stronger inhibiton, is less ADP produced and a lower luminescence
the signals are normalized using
high control: luminescence without inhibitor
low control: luminesence without enzyme (no ADP)
these signals at different concentrations is converted into a dose response curve from which the concentration at 50% inhibiton can be determined = IC50
What are host cellular factors
Host cellular factors are molecules or processes from the host cell that a virus uses to complete its life cycle.
kinases
host enzymes
membrane trafficing proteins
…
does inhibition of kinases cause side effects
Erlotinib is an example of an EGFR (Epidermal growth factor receptor) kinase used to treat and control certain cancers (like lung cancer)
But EFGR is also present in healthy cells in escpecially the skin and gastrointestinal tract. so inhibition of those can cause side effects such as skin rash or diarrhea;
what is a chemical proteomics approach
proteomics is the study of proteins
Chemical proteomics is the combination of chemistry and proteomics to identify proteins targets of small molecules. small molecules are used as probes by putting them on a solid support such as beads which are used to fish out proteins form a complex biological sample, and the proteins are then identified through mass spectrometry. This helps confirm binding to a target or identify off target interaction
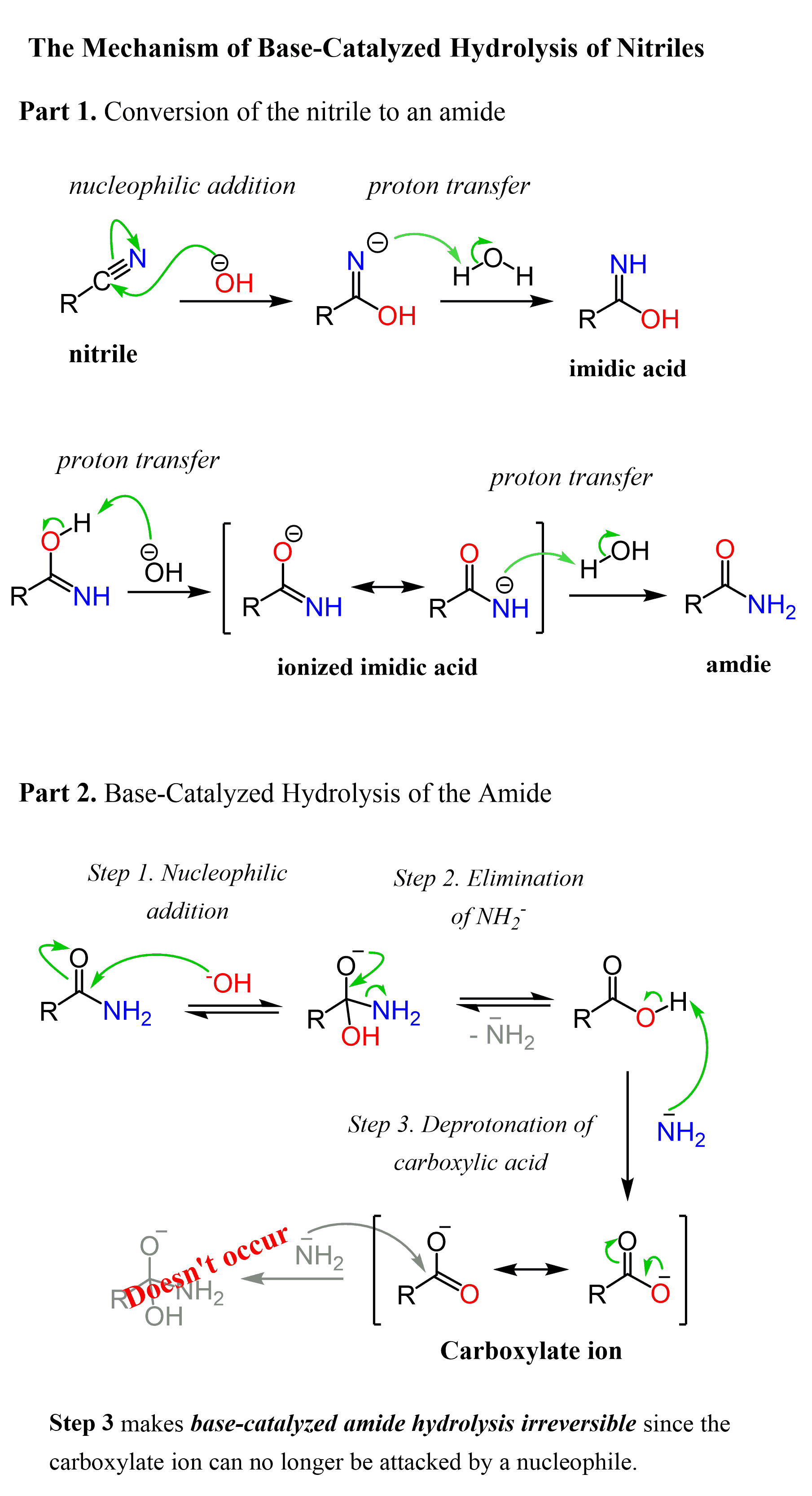
what is Kd, what is it a measure of adn why do lower values mean higher affinity?
what is Ki
Kd = dissociation constant
it gives an indication of the binding affinity.
it is a dissociation constant so Kd is equal to the ligand concentration time the protein concentration over the bound concentration at equilibrium, therefore a higher value means a preference to dissociate, a higher ligand and protein concentration and therefore lower affinity.
Ki = inhibitions constant
also a equilibrium constant like Kd and describes the affinity for the inhibitor for the protein during an inhibition assay, so it describes the inhibition of activity while Kd describes only binding
measurment:
Kd is measured by measuring how much ligand binds to protein at differen ligand concentrations and fit a binding curve
Ki is measured by measuring the enzyme activity at different inhibitor concentrations like dose response ADP-Glo kinase assay
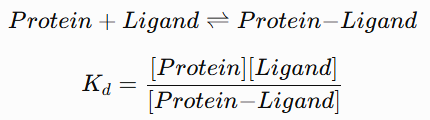
why does alkylation of 12 occur on the 3 position an dnot on 6?
because they are not equally reactive
Br at the three position is attatched on an electron deficient isothiazole aromatic ring close to the S and N making it mor electron deficient and susceptible to nucleophilic attack
the other Br is less reactive and therefor remains for the following suzuki coupling
electrophilic aromatic substitution on pyridine is at the 3 or 5 position
the N accts as an electron withdrawing substituent
substituent of the 3 position allows the distrbution of the positive charge on only carbons and no unfavorable resonance structure with the positive charge on the electronegative N
what is EC50, EC90 CC50 and IC50, how are they a measure of antiviral activity and how do they compare?
EC50 = half maximal effective concentration
concentration at which gives 50% of the maximum antiviral effect
eg reduces viral replication by 50%
EC90 is more strict which is the concentration wich gives 90% of the maxim antiviral effect
IC50 = half maximal inhibitory concentration
concentration that inhibits 50% of a certain activity such as kinase phosphorylation (dose response ADP-Glo kinase assay)
but nothing about the antiviral effect
CC50 = half maximal cytotoxic concentration
concentration of compounds at which 50% of cells are dead or don’t perform as they used to
measures toxicity to cells
what is ON
overnight
what is a chemogenomic screening
combining chemistry and genomics
= a method to find which host genes or cellular pathways are important for the effect of a compound (eg. for viral replication)
this is done by treating the cell with the compound while at the same time altering genes and seeing how they affect the response.
what is nak selectivity, what does it stand for and how do you determine it, explain easily and concise
= how selectively a compound inhibits GAK over other NAK family members like BIKE or AAK1
it is calculated by the ratio of the off target Kd or IC50 of the NAK divided by the Kd or IC50 of GAK
a value of 16000 means it binds to GAK 16,000 times more strongly than it does to other NAK family members
Lipinski’s rule of five
Lipinski’s Rule of Five is a widely used guideline in drug discovery to predict if a chemical compound is likely to be orally active and bioavailable in humans. [1, 2]
A molecule is generally considered "drug-like" if it satisfies the following criteria: [1, 2, 3]
Molecular Weight: ≤ 500 Da (Daltons/g/mol)
Hydrogen Bond Donors: ≤ 5
Hydrogen Bond Acceptors: ≤ 10
Lipophilicity (Log P): ≤ 5 [1, 2, 3]
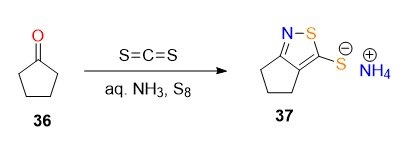
mechanims
Takehsima: Cyclopentanone first forms an enamine with ammonia. This enamine reacts with carbon disulfide to give an aminodithiocarboxylate intermediate. . Finally, under the basic ammoniacal conditions, the thione is present as the ammonium thiolate salt.
The reaction seams to work without S8, but its involvement may have something to do to with stabilising the thiolate or preventing oxidation of the thiolate
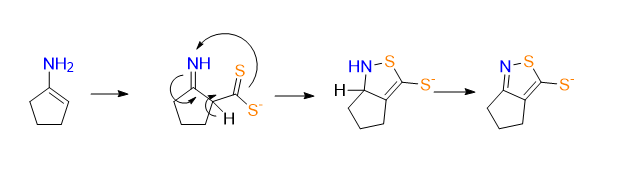
why is the aldehyde signal of the amine so much more shielded?
Local diamagnetic shielding: (isotropic = independent of position in magnetic field)
when a proton has more surrounding electron density, those electrons circulate in an external magnetic field, this creates a small induced magnetic field that opposes the applied field. Therefore the proton fiels a weaker effective magnetic field and apears at lower ppm
as opposed to Cl and the azide who are both electron withdrawing groups (causing deshielding), the amine donates electrondensity through conjugation tho the aldehyde, increasing é density around the aldehyde proton causing shielding
in house optimized conditions at 70 °C, which kind of optimizations were done to warrant this decision?
important because the previous section says there is a disadvantage of using elevated temperatures.
Solvents commonly used for thionation wer screened: toluene, DMF, ACN
ACN obtained the highest yield
The inital thionations during optimization using P4S10 and lawesson’s reagent were reported at 70 °C and therefore this was the starting point of optimization
lowering to 50 °C showed oderate yield loss
25 °C significantly slowed the reaction leading to lower yields
all 4 of these were conducted for 4 hours
next move on to only testing 70 °C using P4S10 with pyridine which performed the best for 20 h
later the pyridine complex was isolated, performed better and its equivalents were varied at 70 °C in ACN and settled for 1 eq.
—> not ideal optimizaton
This temperature is likely need to thermally cleave the thioaldehyde trimers that readily form at room temperature allowing them to ringclose
indeed this is not optimal for the azide degradation, these finding were found only after this optimization, so my proposal:
allow the thionation to run at room temperature for certain amount of time and minimize nitrogen extrusion without ringclosure, and then increase temperature to break up the trimers and enable a milder ringclosure with minimal azide degradaton
For some reaction you observe a non-formylated 4-chloro-1,2-dihydronaphtalenecarbaldehyde side product, i’m confused which product you are referring to exactly?
Yes I understand your confusion, this was a bad choic of wording, the final carbaldehyde shoul be be removed, i’m refering to 1,2-dihydronapthalene so without the aldehyde.
So normally the vislmeir reaction proceeds by the enol of the tetralone to attack on the vilsmeier reagent that is eventually transformed into the aldehyde.
but due to the metoxy group, possibly even due to delocalization, the oxygen of the keton because more electron rich an can attack on the vilsmeier reagent (or left over phosphory chloride, this creates a good leaving group and can be substitued by the choride that was liberated through this process. the nucleophilic character of the compound is now diminished and no aldehyde can be introduced anymore.
this happened to a large extent, for the scale up of 1.5 g even 50 % was this non formylated product, 40 % the desired product and 10% unreacted starting material or degradation, or other side reactions,…
for the fromation of the tetrahydropyranone, why increase the temperature to 50 °C only for the final hour;
this was actually not reported , they only used a 2hour reaction time at room temperature, we improvised to increase temperature for another hour because we didn’t see a full conversion and then stopped the reaction
on TLC we saw two spots that were different from the starting material, so side product formation and one was product, this was maybe the reason why switching to room temperature was a better idea
The only thing that was based on literature was the 2 hour reaction time.
the side products are still a little unclear, it wasn’t any aldol condensation with itself or with DMF, so still unclear
possible explanation:
the procut isn’t as stable as it is for the tetralones or acetophenones through conjugation, meaning that the transitions state of the tetrahydropyranones is higher in energy making the reaciton more difficult
also protonation of the oxygen could lead to side reactions
might also have something to do with the geometry of the 6 memberad ring going from chair to half chair
the yield of product teterahydrthiopyranene is only 18%, so why was it conducted for a shorter duriation and why didn’t you increase the temperature like you did with teterhydropyranone?
since we didn’t observe an additional conversion after increasing the temperature for the final hour, we decided to just follow the reported procedure since the scaffold is very similar to the oxygen one. After two hours we already saw the starting material was completely consumed and one larger spot was present ‘the product) and one baseline spot
the extraction of this compoudn was tedious, so a lot of water remained in the organic phase therefore a lot of MgSO4 need to be added for drying, so we likely lost a lot product there and also during column all tubes were clean, so additional losses
we still obtained enough compound to continue so we didn’t look into it further, optimization also wasn’t the goal of the project. It was just quickly seeing if they were synthetically accessible for them to be included in the virtual screeninng process. Lookin back on it now, i think this could be easily otpimized to above 25% yield and could be included for the screening process
Increased yield of the thio variant
It’s hard to explain, it could have something to do with if the oxygen protonation leads to side reaction, because this is less likely for sulfur and also that sulfur is larger therefor a slightly alternative geometry of the 6 membered ring
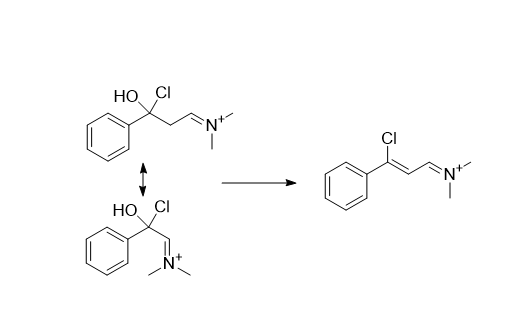
is there an explanation why the Z-isomer is formed in large excess?
Following the reaction mechanism, after the addition to the vislmeier reagent, the cloride attacks on the ketone and expels water creating the double bond, as this dimethylammonium group is pretty large it prefers to be trans to the large phenyl group due to steric hinderance, so it is more likely the compound is in the this configuration before the double bond is formed.w
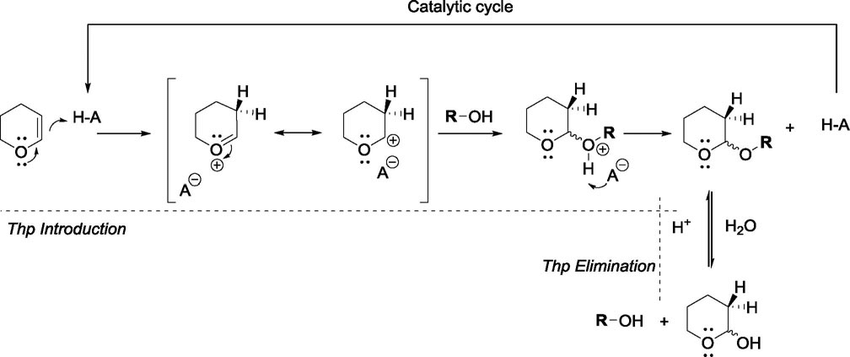
what is the mecahnism of THP protection
protonation activates the double bond
this creates an oxocarbenium ion stabilized by oxygen
the alcohol attacks at the electrophilic carbon
deprotonation gives the THP protected alcohol
how are the protection groups deprotected?
THP
adding water (acidic aqueous conditions)
the alcohol group is protonated, water attacks liberating the alcohol
TBDMS
strong acid or fluoride (like teterabutylammonium fluoride = Bu4N+F-) fluoride has high affintiy for Si
difference between these terms: potency, activity, selectivity and affinity
affinity
how strongly an inhibitor binds to the target
potency
how much inhibitor is needed to induce a certain effect (EC50 or IC50)
activity
more general term, telling us if the compound prodcues the the desired effect (%inhbition EC50 or IC50)
could include kinase inhibition but also influence on viral replication
selectivity
preference for a target
represented using selectivity ratios of IC50 or Kd