2005 wk 8 lec
1/63
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
64 Terms

Histamine chemical structure
Synthesised via histidine
Histidine (amino acid) contains carboxylate group
Decarboxylation occurs (via HDC)
Formation of Histamine
Can come from diet and foods as well eg. wine
Where is histamine found?
Mast cells
Basophils (in blood)
The lungs, GIT, and skin have the highest no. of mast cells
Hence the reason for most allergic reactions occurring at these sites ?
How does Histamine act?
Acts as an agonist to different Histamine receptors
eg. H1, H2, H3, H4 receptors (all of which are G-protein coupled receptors)
These H receptors have constitutive activity
Meaning they can be activated and inactivated regardless of Histamine binding (eg. can initiate contraction of lungs without histamine)
Why are antihistamines called antihistamines instead of antagonists?
They are classified as inverse agonists
Inhibit histamine from activating the receptor
Even though they block the H1 receptors, they cannot be classified as antagonists as they are acting on the constitutive activity
They stabilise and preserve the inactive form, so even though histamine can still bind → it cannot trigger an action
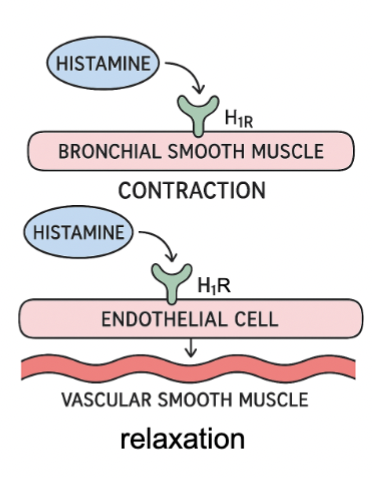
Stimulation of H1 receptors
Directly (eg. on bronchial smooth muscle)
Contraction of smooth muscle
eg. bronchoconstriction
Indirectly (eg. vascular smooth muscle / blood vessel)
Relaxation (vasodilation)
Release of Nitric Oxide which induces relaxation of VSM
Itchiness
As sensory neurons become increasingly sensitive
CNS arousal and awakening (feeling awake
Which is why 1st gen antihistamines cause drowsiness and feeling sleepy
Histamine Triple Response
An experiment to observe whether scratching the skin would stimulate the same response as injecting histamine locally
Both of which caused:
Redness of skin
Surrounding flare of skin (beyond the scratch site redness - spreading a little further out)
Weal (the slight bump)
Conclusion: Scratching replicates the stimulation of H1 receptors

Type 1 Hypersensitivity
IgE Allergic reaction
Occurs when a harmless substance is mistaken by the immune system as a threat
Lack of tolerance leads to sensitivity
Prior exposure to the allergen causes the immune system to produce IgE antibodies against it
IgE antibodies bind to mast cells and basophils, ready to initiate an immune response for future exposure
Majority of people do not react or produce sensitivity to these allergens, but some may develop it
Examples of Allergens =
- Peanuts
- Pollen
- Seafood
- Penicillin
Important to note: there are many additional mediators to allergic reactions eg. which is why antihistamines are not first line treatment for asthma due to other contributing factors (eg. Mast cells, basophils, histamines + more)
Sensitisation TH2 driven process *****
TH2 = Type 2 Helper T cells ?
Antigen presentation to Antigen Presenting Cell (APC)
Consumed by APC
APC binds to TH2 cell
IgE production
Mast cell degranulate and release cytokines (this does not occur immediately; but rather following the initial exposure to the allergen, it is prepared for the next exposure)
Cytokines = trigger for TH2, Histamines is the trigger for Type 1
→ Clinical effects:
- Asthma
- Eczema
- Anaphylaxis
- Allergic Rhinitis (Hay fever)
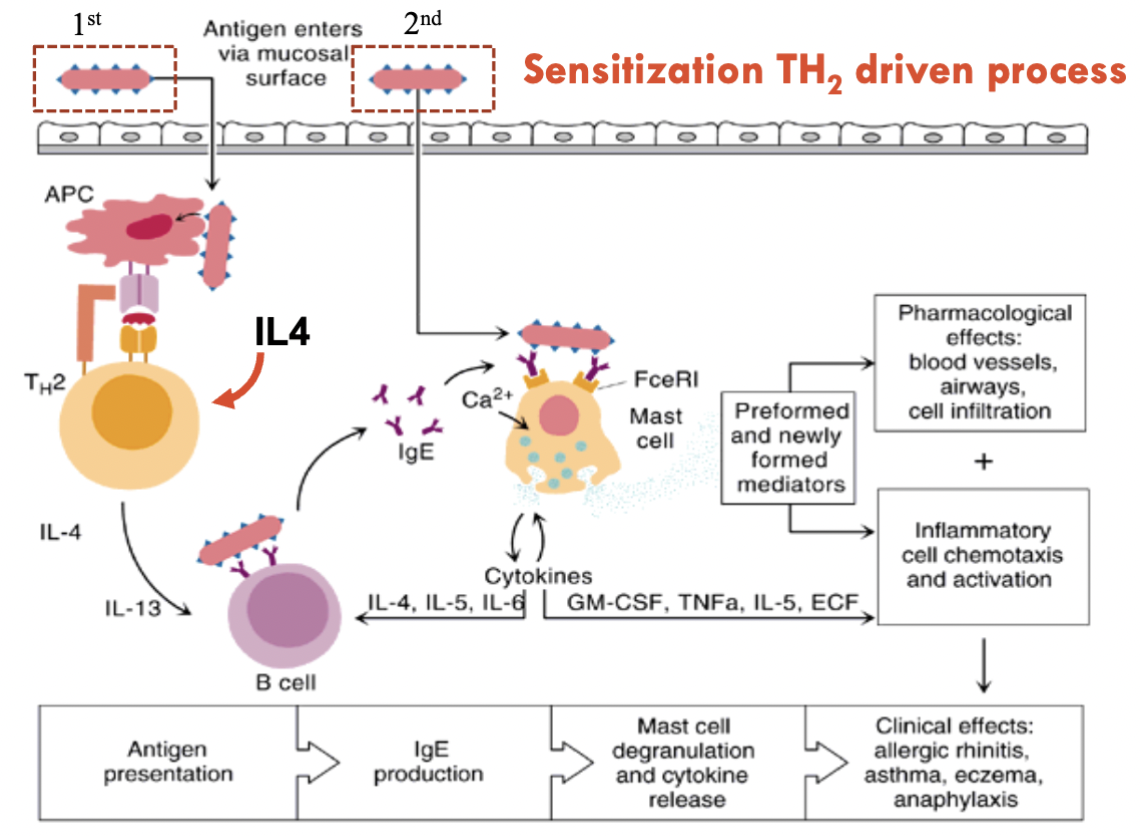
IgE receptor activation
Antigen binds to IgE antibodies
Once antigen binds, the antibodies must cross-link
Degranulation is triggered and initiates release of cytokines
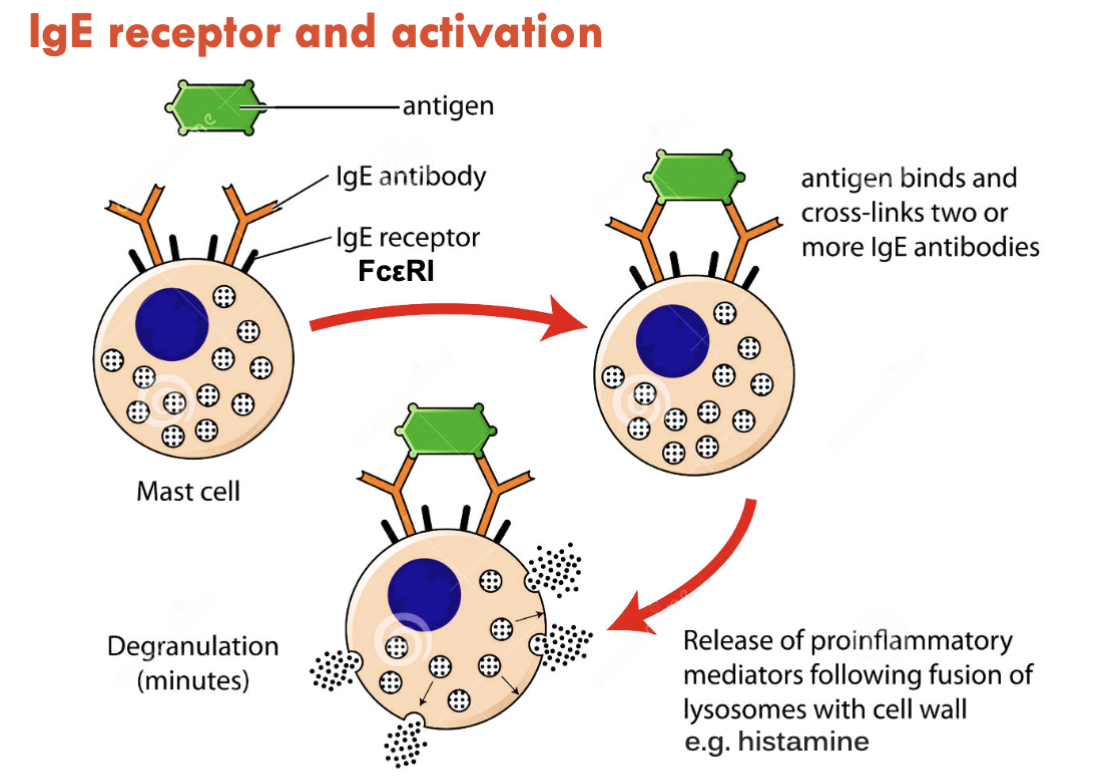
Non-IgE receptor mast cell activation
Pseudoallergic reaction
Can occur on first exposure (as opposed to IgE mediated = first exposure triggers hypersensitivity for the next exposure)
Mas-related G protein-coupled receptors:
Morphine
Vitamin K1
Substance P
Allergic Rhinitis
Histamine release from mast cells stimulate H1 receptors to produce:
Vasodilation
Itchy, swollen red eyes
Itchiness
Runny noseSneezing
Skin rash and Hives
Histamine release from mast cells stimulate H1 receptors to produce:
Vasodilation
Skin rashes
Weals
Itchiness
Redness
First-line for Allergic Reactions
F.A.S.T
Face
Rash
Swelling of lips
Eyes
Airway
Breathing difficulties
Swallowing
Speaking
Stomach
Abdo pain
Vomiting
Diarrhoea
Total Body
Rash
Swelling
Weakness
Role of Epinephrine in Anaphylaxis
MOA:
Alpha Agonist → Vasoconstriction, reverse hypotension and reduce oedema
B1 Agonist → Increase Cardiac output
B2 Agonist → Bronchodilation
Use of Epipen:
Use ASAP to reduce mortality risk
Injected into mid-thigh - slow release
Antihistamines
Inverse agonists
Act on H1 receptors and induce inactive state of constitutive activity
H2 receptor Antagonists
eg. Famotidine
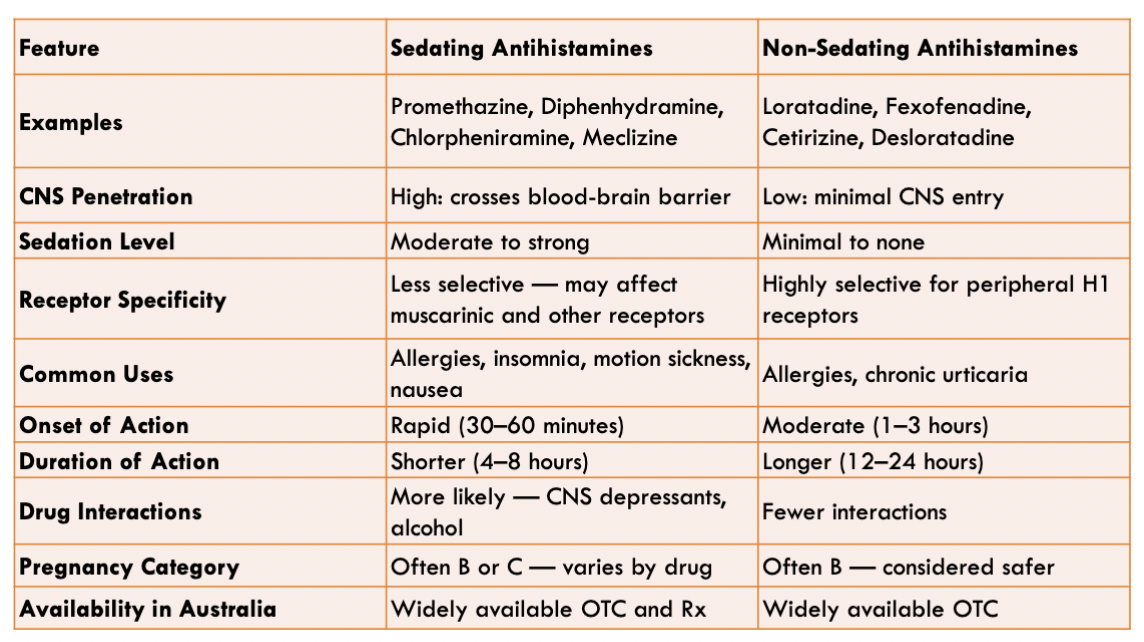
Sedating Antihistamines
Promethazine (allergies)
Doxylamine (Sleep Aid)
Sedation is caused when these drugs cross BBB acting on the CNS H1 receptors → causing sedation
Therapeutic Uses:
Allergies
Insomnia
Motion Sickness
→ be ware they may induce antimuscarinic effects due to non-selectivity and action in the CNS
Effects can last from 8 - 30hrs
SEs of Sedating Antihistamines
CNS depression
Should not be combined with benzo’s, opioids, alcohol etc. due to antimuscarinic effects)
High drug interactions risk
Treatment of Motion Sickness
Dimenhydrinate
Diphenhydramine
H1 antihistamine and anticholinergic effect
Hay fever treatment ****
Non-sedating Antihistamines
Loratadine (Claratyne)
Cetirizine (Zyrtec) → Slightly more sedating than Loratadine
Longer half lives
More selective for H1 receptors
Used for allergic rhinitis etc.
Can be used with CNS depressants as opposed to sedating Antihistamines
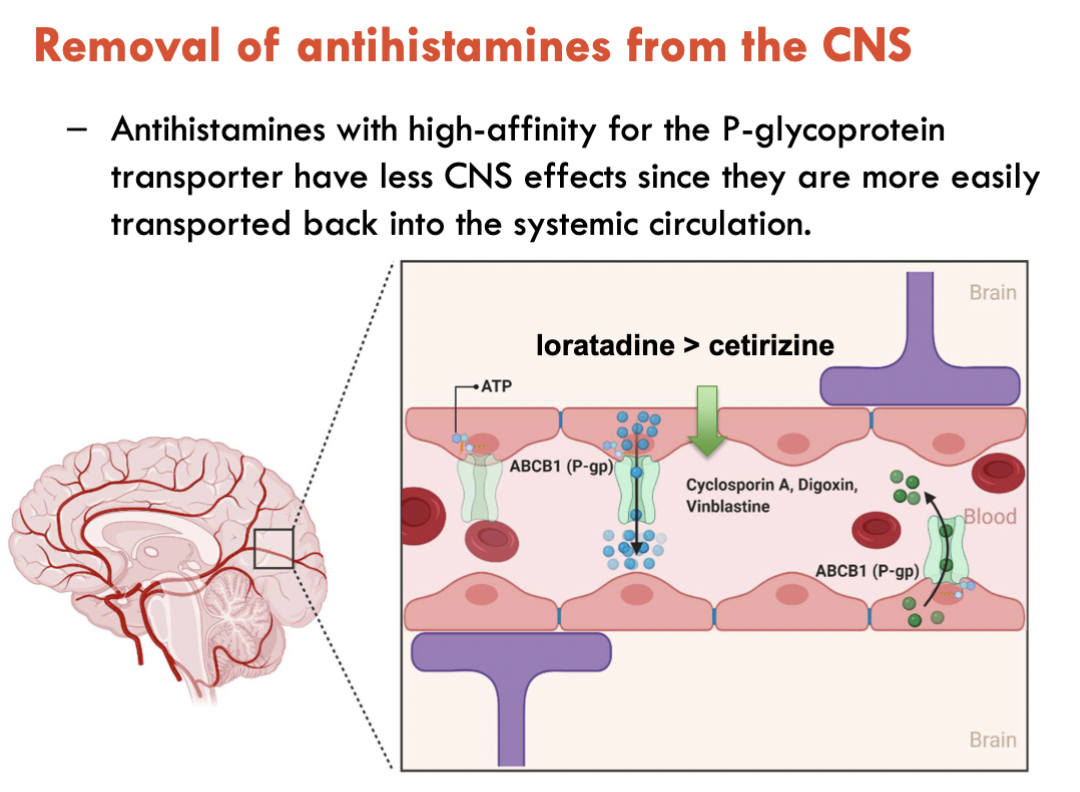
Antihistamine transport from CNS
Antihistamines have high affinity for P-glycoprotein transporter
Easily transported from CNS to systemic blood circulation
Loratadine > Cetirizine for affinity
Green = P-glycoprotein transporter
If antihistamine remained in CNS, histamine would continue to be antagonised → Inducing drowsiness and sleepiness as blocking histamine blocks the promoting of wakefulness
Sedating vs Non-sedating antihistamines

Why do we use NSAIDs / Paracetamol?
Pain
Inflammation
Mediators that induce pain:
Prostaglandins
Leukotrienes
Substance P
Bradykinin
NSAID examples
Traditional:
Aspirin (> 300mg)
Ibuprofen
Naproxen
Diclofenac
COX-2 inhibitors:
Celecoxib
Meloxicam
Use of NSAIDs
Back aches, headaches
Muscle aches and pain
Gout
OA / RA
etc.
How do NSAIDs do?
Analgesic (decrease pain)
Anti-inflammatory (deactivates cells)
Antipyretic (decrease heat)
Antiplatelet
By blocking the production of prostaglandins through the inhibition of COX enzymes
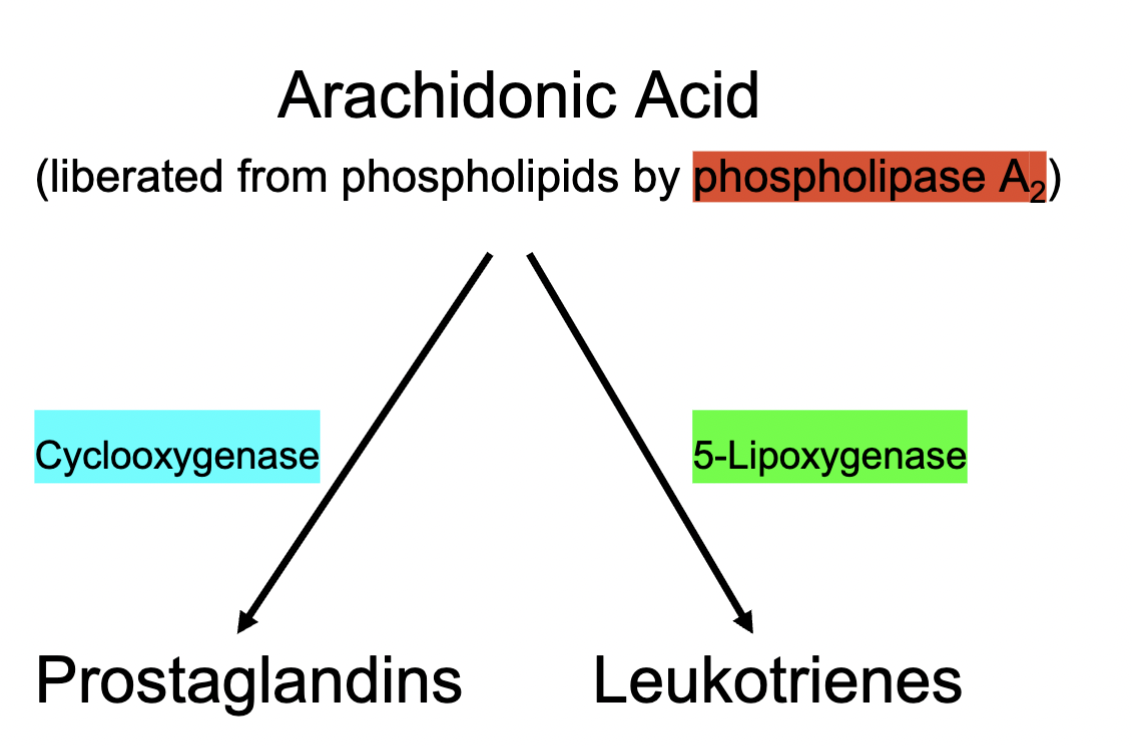
Production of Prostaglandins and Leukotrienes
Phospholipase A2 activated
Generates Arachidonic Acid
Arachidonic acid becomes metabolised through many pathways; the main two being
COX → Prostaglandins
5-Lipoxygenase → Leukotrienes
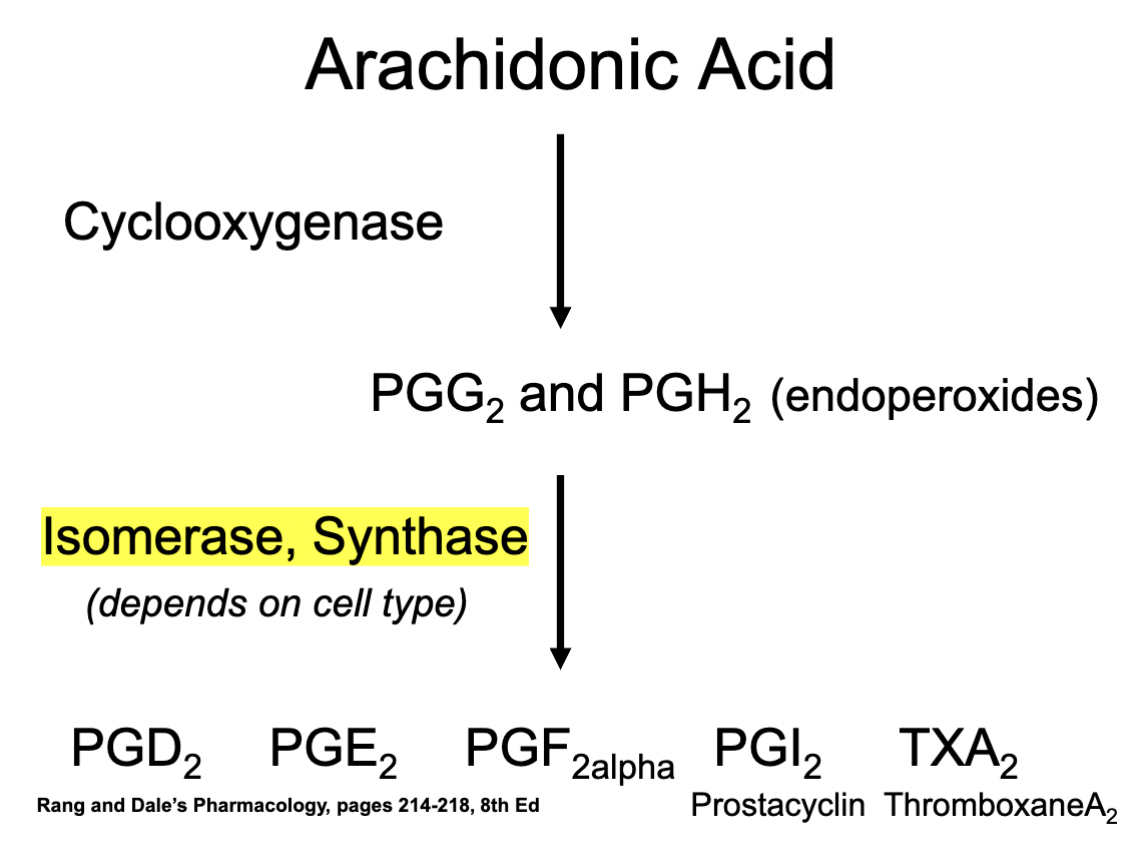
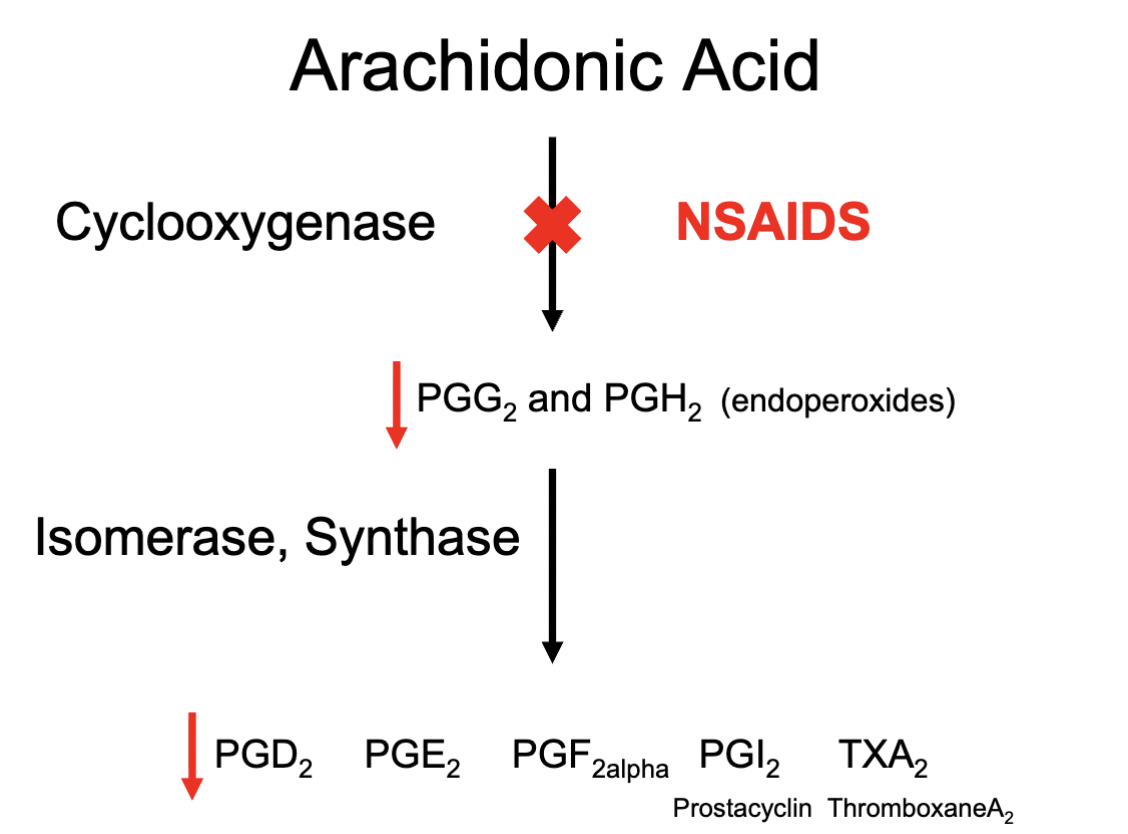
Eg. of production of Prostaglandins through COX
1) Arachidonic Acid
2) COX metabolism
3) PGG2 & PGH2
4) Production of Prostaglandin types through Isomerase / Synthase
Depends on the cells type and what type of the enzymes are available
5) Formation of different types of prostaglandins (and their respective subtypes)
How do NSAIDs work?
Blocking COX enzyme responsible for metabolism of Arachidonic Acid; reducing production of prostaglandins
COX-1 Enzymes
Found in most cells
Constitutive (always making) enzyme that synthesises production of prostaglandins involved in homeostasis
AKA the “good” prostaglandins
COX 2 Enzymes
Induced by inflammatory response
Synthesises prostaglandins in response to pain and inflammation
Considered the “bad” prostaglandins
SOME COX 2 enzymes are constitutive (initially thought to always be induced by pain / inflammation)
eg. in the kidneys, vascular tissue
Therefore important to be wary of inhibiting to an extent through NSAIDs
What do prostaglandins do?
Act on G-protein coupled receptors
Eg.
PGE2
PGI2
Involved in pain and inflammation
Induce vasodilation
Increase permeability of blood vessels
Increase sensitivity of nerves to pain stimuli (They do not directly produce the pain; but rather increase the sensitivity and detection to minor stimuli inclusive)
Traditional NSAIDs MOA
Block the production of ALL prostaglandins
Inhibition of both COX 1 + COX 2
Why is this important?
It can provide beneficial therapeutic effects by blocking COX-2 induced pain and inflammation
Analgesic & anti-inflammatory effect
Inhibition of COX 1 can induce potential ADEs and disruption to homeostasis
Example of “housekeeping” / “good” prostaglandins
Those that help maintain mucosal layer in stomach
Those that reduce gastric acid secretion
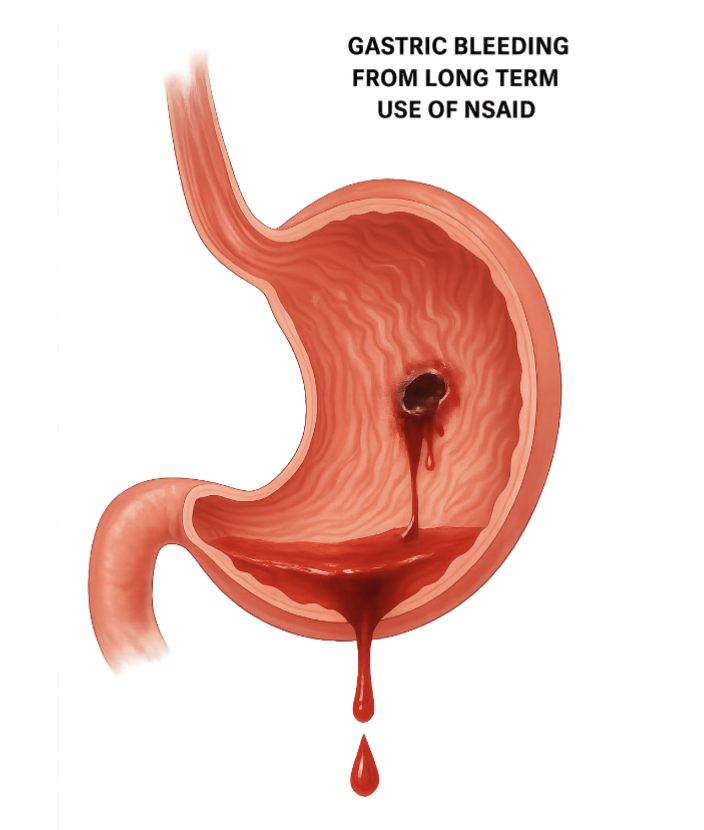
Stomach and NSAIDs
Highly acidic pH
Pepsin (digestive enzyme)
NSAIDs may reduce the protective gastric mucosal barrier which in turn exposes the stomach lining to highly acidic and dangerous fluids and enzymes that can cause PUD
What does COX 1 do for the stomach?
Increase bicarbonate secretion
Increase mucosal blood flow
Increase mucous secretions
Reduce gastric acid secretion
Another example of “Housekeeping” Prostaglandinds
COX 1 that regulates platelet function
Eg. TXA2, PGI2
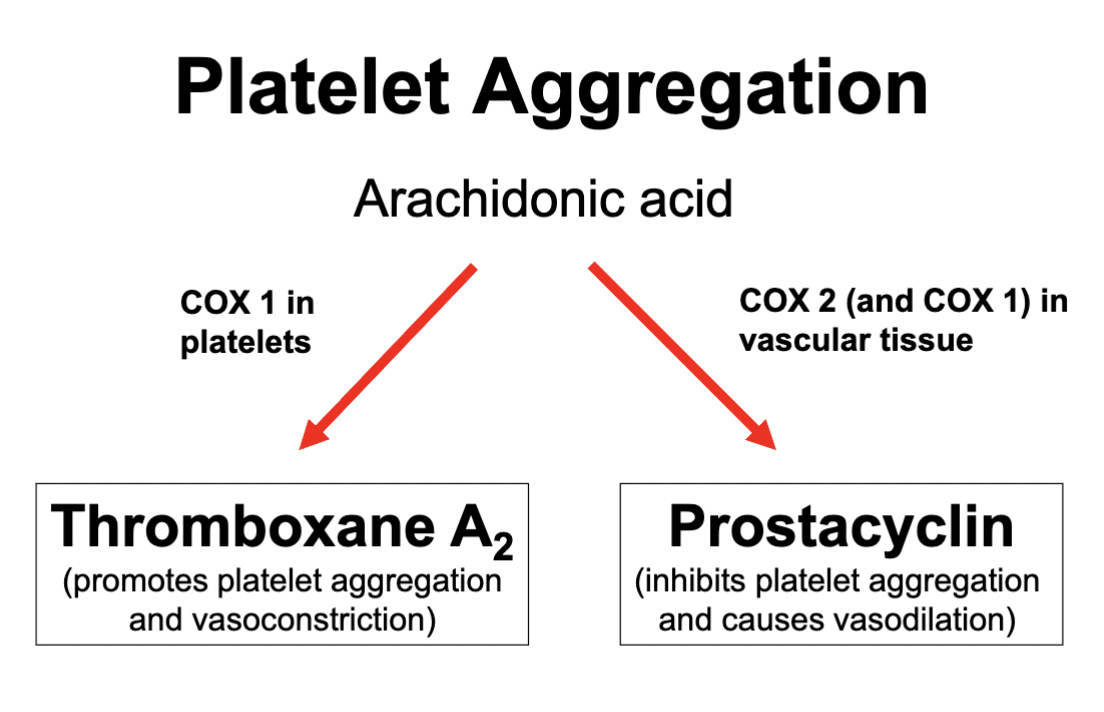
TXA2 & PGI2*****
Thromboxane is formed in platelets by COX 1
Induces platelet aggregation
Produces vasoconstriction
Contributing to clotting factors
Prostacyclin is formed in the endothelial cells by COX 1 and COX 2
Inhibit platelet aggregation
Vasodilation
COX 1 / COX 2 in platelet aggregation
Thromboxane = COX 1 produced
Promotes platelet aggregation
Vasoconstriction
Prostacyclin = COX 2 and COX 1 produced
Inhibits platelet aggregation
Vasodilation
These 2 balance each other out by inducing opposite effects in order to control platelet aggregation
Low dose Aspirin
Inhibits COX 1 in platelets → inhibiting TXA2 production
Which in turn reduces platelet aggregation
Reduces the effects of vasoconstriction
Aspirin binds irreversibly to COX 1 - meaning COX 1 in platelets cannot regenerate in comparison to COX 1 & 2 in vascular tissue (which can regenerate)
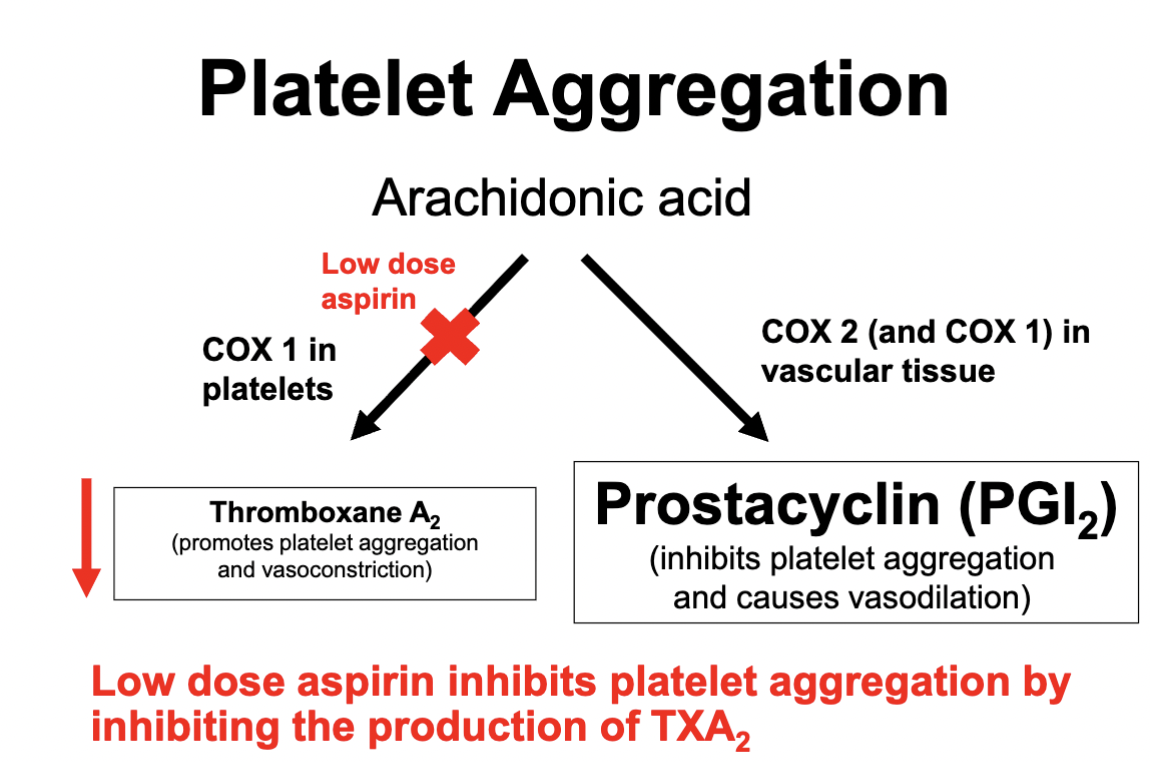
How does lose Aspirin work?
Background info:
COX 1 converts Arachidonic Acid in platelets into Prostaglandin Thromboxane A2 (TXA2)
TXA2 induces platelet aggregation and vasoconstriction
Now what does Aspirin do?
Irreversibly binds to COX 1
COX 1 in platelets cannot regenerate - therefore binding to COX 1 in platelets allows for the reduction in platelet aggregation and vasoconstriction
Vascular Tissue can regenerate COX 1 & COX 2 - therefore inhibiting it would not induce as much of a therapeutic effect
More “Housekeeping” Prostaglandins
Those that help maintain renal function
eg. PGI2, PGE2 (COX 2)
Those that help airway function in patients with asthma
Eg. PGE2
+ Assist implantation of fertilised ovum
ADEs of NSAIDs
GIT bleeding and PUD
Reduced renal function
Na + water retention
Miscarriage
Asthma AE
Bronchoconstriction
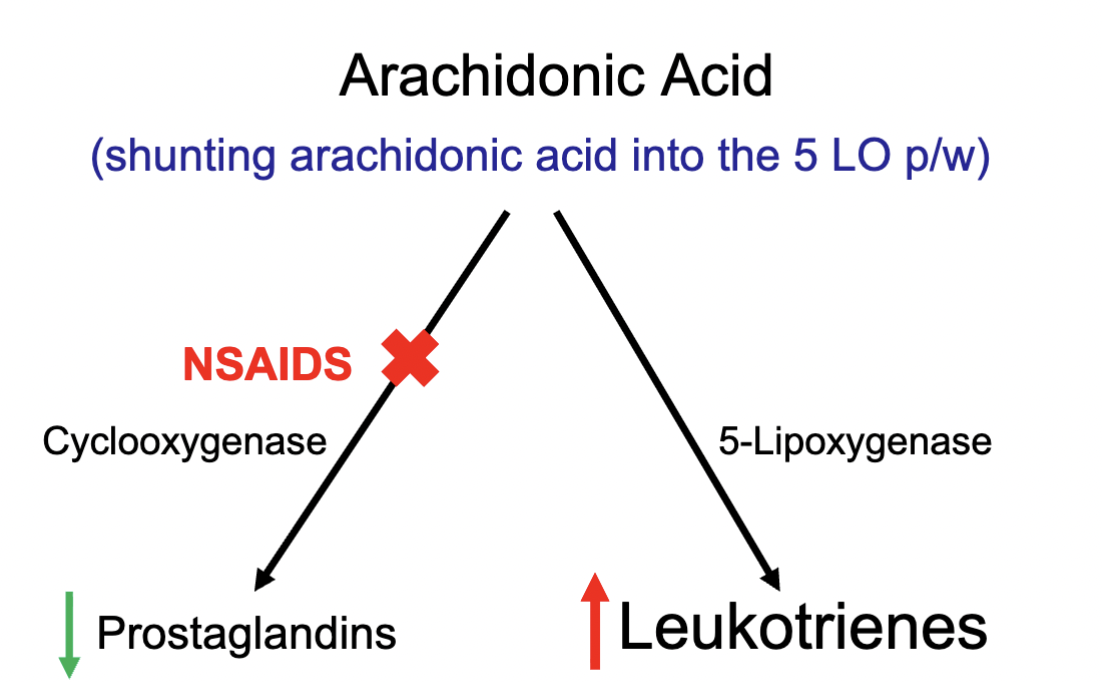
NSAIDs in asthma / lung function
When using NSAIDs, Arachidonic acid cannot be metabolised via COX as the pathway is blocked
Decrease of prostaglandins
However, the AA has to metabolise in some form, therefore Leukotrienes conc increases
Leukotrienes promotes narrowing of the airways (bronchoconstriction)
NSAIDs for elderly patients
More susceptible to ADEs of NSAIDs due to reduced renal clearance
Use of NSAIDs with caution; why?
Potential PUD
Cardiac failure
Renal failure
HTN
Asthma
Pregnancy
Antipyretic effect of NSAIDs
Inhibition of prostaglandins in the hypothalamus
Only reduces elevated temperatures - NOT regular body temp.
Drug interactions of NSAIDs
ACE i
ARBs
Diuretics
Warfarin
Methotrexate
COX 2 inhibitors (selective ?)
Celecoxib
Meloxicam
Produce the same analgesic and anti-inflammatory effect as traditional NSAIDs
Benefits of COX 2 i
Produce less GI bleeding and ulcers ADEs
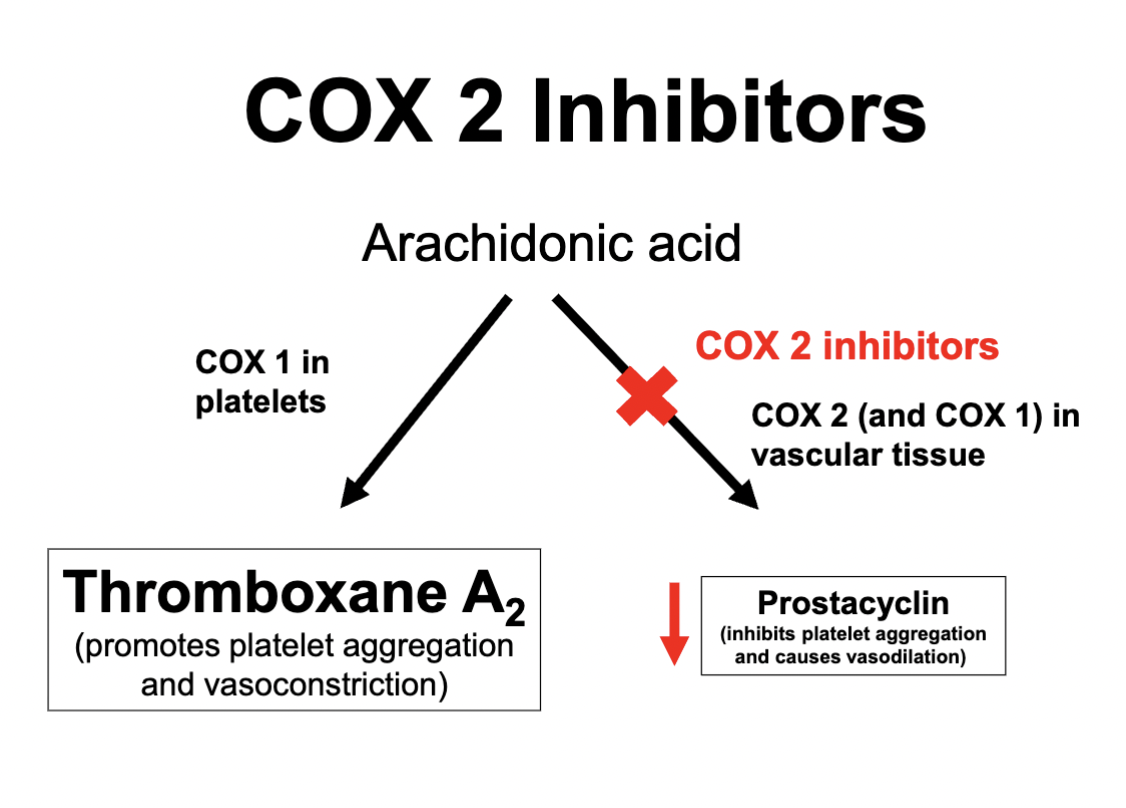
Do not inhibit platelet aggregation and vasoconstriction
As TXA2 is responsible for this; via COX 1
ADEs of COX 2 selective NSAIDs
Renal effects
Use with caution in renal impairment
Use with caution in cardiac failure
HTN exacerbation
Stroke
This is because inhibiting COX 2 reduces prostacyclin conc, which leaves elevated levels of TXA2. When TXA2 levels are elevated and there is an imbalance, platelet aggregation and vasoconstriction is increased
Drug interactions of COX 2 inhibitor NSAIDs
ACE i
ARBs
Diuretics
Precautions to consider when using NSAIDs
High doses and term of use
CVD risk
Stroke risk
Renal function
Aim to use for shorter durations and lower doses where possible; this includes ALL NSAIDs
Paracetamol
Max dose: 4g / 24hrs → Typically well tolerated
Analgesic effect
Antipyretic effect
NO anti-inflammatory effects
MOA = not fully understood
When to use Paracetamol
When NSAIDs are contraindicated
CVD
Renal impairment
Severe asthma
HTN
Previous ACS
Elderly (eg. OA where NSAIDs are necessary)
Benefits of of using Paracetamol in comparison to NSAIDs
Fewer drug interactions
Does not induce PUD or GI bleeding
No exacerbation of renal function
No effects of platelet aggregation
ADEs of Paracetamol
Liver toxicity and metabolic ADEs
Over production of NABQI via CYP450 enzymes
NABQI is toxic; though typically conjugated with glutothione in an inactivated form
What happens when Paracetamol overdose occurs?
As glutathione is typically conjugated with NABQI, Glutathione stores become depleted
When there is no more Glutathione, NABQI becomes activated
NABQI on its own can bind and with other cell components - causing liver toxicity and death
Symptoms of Paracetamol OD
N / V (initial symptoms)
Serious liver damage symptoms:
Jaundice
Metabolic disturbances
Although, typically undetectable until lab tests eg. LFT
Treatment of Paracetamol OD
Restore Glutathione levels
Provide IV Acetylcysteine
Goal is to deactivate NABQI → Aim to treat within 12hrs post-OD