Cancer cells: reduced death rate
1/17
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
18 Terms
… rates are high in tumours because the cell environment is…
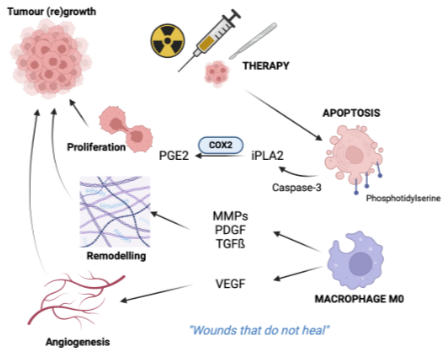
Apoptosis is high due to inhospitable and cell signalling is dysregulated. Apoptotic cells stimulate wound healing and responses that promote tumour groth and regrowth after therapy
High cancer cell mitosis rates are balanced by…
Programmed cell death
although lytic necrotic cell death can also occurs, large sale normally “tidy“ apoptosis can trigger inflammation and wound healing
Apoptosis can occur at any point in the tumour development to promote the initial tumor growth or regrowth after cytotoxic therapy
Apoptosis promotes wound healing
Effector caspase-3 cleaves phosphoplipase A (iPLA) from cell membrane lipids, which produces aranchodionic acid which is metabolised by COX2 into PGE2 which promote the increase in vasodilation for cancer cell proliferation
Apoptotic bodies are coated with phosphatidylserine (PS) which isan eat me signal for macrophage to come and ingest which then release
VEGF → angiogenesis
MMPs, PDGR, TGFb → extracellular matrix remodelling/ fibrosis
Suppression of apoptosis
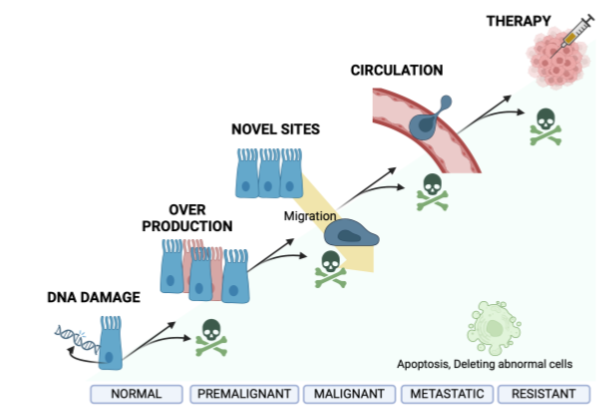
Individual malignant cells can suppress apoptosis, which is important for the proliferation of cancer cells from initiation → progression → treatment resistance
Suppression of apoptosis at the premalignant stage
Extend lifetime of cells so allow accumulation of potential “driver” mutation
Alters normal tissue homeostasis resulting in over production of cells (hyperplasia)
Supression of apoptosis pre malgnant to malignancy
Allow suppression of anoikis - cells can survive without attachment to the basement membrane
Cells can therefore move to invade tissue -metastasis
From metastatic to resistant: advantage from suppression of apoptosis
Allow cells to survive in the inhospitable environment (full of pro-apoptotic signals from immune cells eg FasL)
Allow cells to be resistant to therapies such as hormonal treatments that are used to kill cancer cells
Stages of cancer cell
Normal - Premglinant - Maglinant - Metastatic - Resistant
What are the two apoptosis pathways
Mitochondrial (intrinsic pathway) or death receptor (extrinsic pathway)
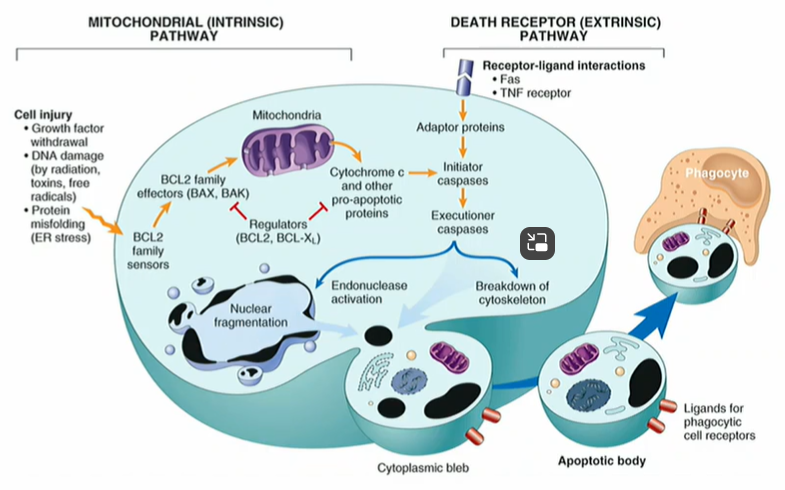
Apoptosis by extrinsic pathway is driven by
External signals detected by cell surface receptors eg Fas receptor
Fas receptor binds to FasL (Fas ligand)
Fas ligand binds an intracellular adaptor molecule FADD via a ‘death domain’then activates an initiator caspase
The initiator caspase then activates effector caspase which cleaves target proteins triggering the cell apoptosis process
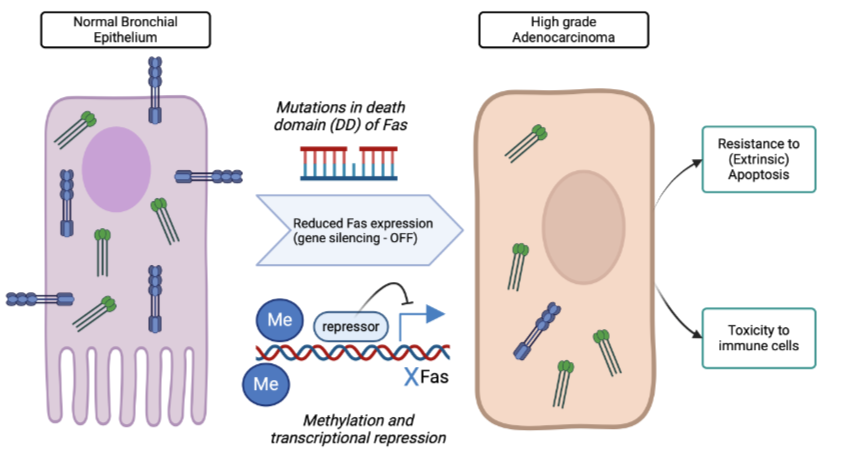
Switching off death receptor pathways in cancer
Fas receptor and FADD can be switched off by mutations in the death domain (inactivated protein) or through promoter methylation (less transcription)
Initiator caspase expression can also be switched off in cancers methylation
This leads to resistance to pro-apoptotic signal
Death of tumour infiltrating lymphocyte cells by secretion of FasL from resistance cancer cells (counter-attack)
Intrinsic apoptosis - BCL2, Bax
intrinsic apoptosis is balanced by pro and anti apoptotic proteins which sit on the outside membrane of mitochondria
BCL-2 is a key sensor of cell stress, it is an ANTI APOPTOTIC protein that is OVEREXPRESSED by chroosome translocacation to the PGH locus in B cell follicular lymphoma, this turns it into an ocogene which extends the life of B cells but does not affect the cell cycle (proliferation rate)
BCL-2 and Bax regulate the movement of pro-apoptotic proteins like Cytochrom C from mitochondria so they do not interact with and activate the APAF-1 apoptosome
BCL-2 closes mitochon membrane pores (no release of CytC, anti-apoptotic_
Bax opens membrane pore (release of CytC, pro-apoptotic)
Oncogenic, anti apoptotic BCL-2 is over expressed in 50% of all cancers
Activating mutant EGFR protein (delEx2-7) can also induce anti-apoptotic BCL-XL expression (as well as activating the cell cycle) leading to treatment resistance
Pro-apoptotic Bax, a tumor supressor gene, is inactivated in colon and stomach cancers
The master regulator - p53 protein (which is encoded by TP53 gene)
Is the most inactivated protein in cancer (it is a tumor suppressor gene)
It is mutated in families which inhereited Li Fraumeni cancer syndrome
It normally regulated apoptosis (pro-apoptotic) and IS A CELL CYCLE CHECK POINT PROTEIN) upon detection of DNA damage (an adaptive stress response to genotoxic damage)
p53 is normally kept in check by
MDM2, which binds and target it for destruction
p53 regulates itself by triggering the expression of MDM2 in a feed-back loop, keeping its activity under control once the emergency stress is resolved
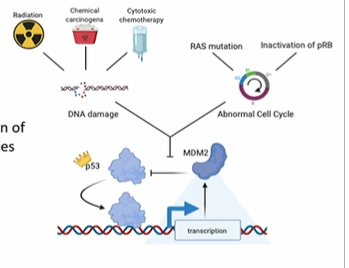
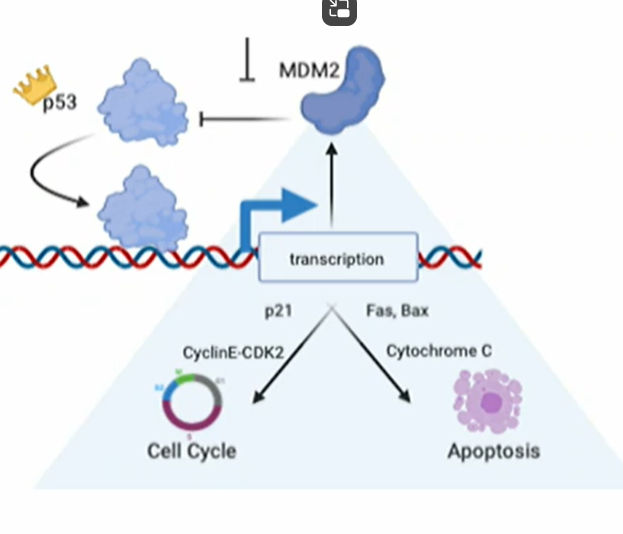
Activated p53 normally induces transcription of proteins that
ARRESTS the cell cycle at G1 by inducing expression of p21cip1, an inihibitor which represses Cyclin E-CDK2 hence allow DNA repair
Failure of repair or excessive DNA damage promotes apoptosis via pro-apoptoxic Fas and Bax (→ cytochrom C release from mitochon)
p53 in cancer (5 effects)
Deleted/ inactivated p53
Gene amplified MDM2
=?
Deleted/ inactivated p53: p53 dependent genes are no longer transcriptionally activated
Gene amplified MDM2: too much MDM2 protein targets p53 for destruction
= no cell cycle arrest = no DNA repair = No apoptosis
If cells survive:
Acccumulation of mutation (DNA damage not repaired)
Proliferation of cells (loss of cell cycle checkpoint)
Malignant transformation
Expansion of the tumour mass
Chemotherapy can select for cells with p53 mutations that are resistant to apoptosis
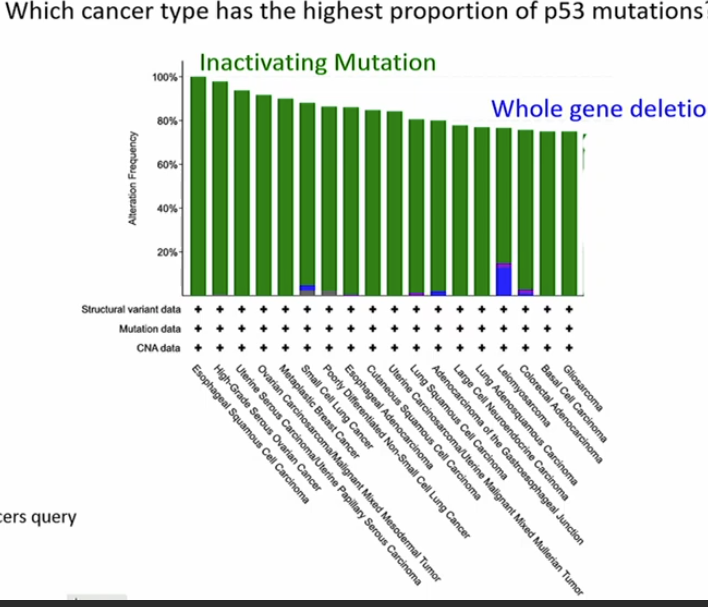
Which cancer type has the highest proportion of p53 mutation?
Esophageal Squamous Cell carcinoma
inactivating mutation - gene product having no function
Why is tumour growth supported by increased rates of apoptosis and decreased sensitivity to apoptosis? Describe mechanisms by which apoptosis is perturbed in cancer cells.