DNA Manipulation
1/53
Earn XP
Description and Tags
Year 12 Biology U3 AOS 1
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
54 Terms
Enzymes that manipulate DNA
Endonucleases → cuts DNA (scissors)
Ligases → joins DNA (glue)
Polymerases → amplify DNA (multiply)
Endonucleases
enzymes that break bonds between nucleotides of nucleic acids
Sticky ends
staggered cuts through a double-stranded DNA, resulting in overhanging nucleotides
Called “sticky” as unpaired nucleotides will be attracted to a complementary set of unpaired nucleotides
Ensures an inserted gene is oriented correctly
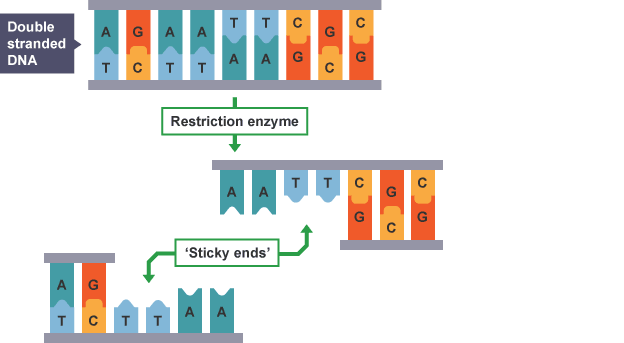
Blunt ends
straight cuts through a double-stranded DNA, resulting in no overhanging nucleotides
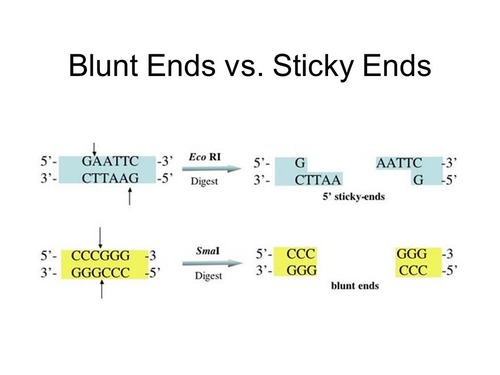
Restriction enzymes
a type of endonuclease that are produced by bacteria + cut DNA at specific recognition sites
Restriction enzymes vs. Cas9
Restriction enzymes — only act on specific restriction site which cannot be programmed
Cas9 — versatile so it can be programmed to target ANY specific sequence dictated by a piece of gRNA
Ligases
enzymes that join nucleic acid fragments together by creating bonds between a sugar and a phosphate (backbone)
DNA ligase → joins two DNA fragments
RNA ligase → joins two RNA fragments
Polymerase
enzyme that synthesises a polymer from monomers, such as forming a DNA strand from nucleic acids
Requires a primer → a short, single strand of nucleic acids that acts as a starting point for polymerase enzymes to attach
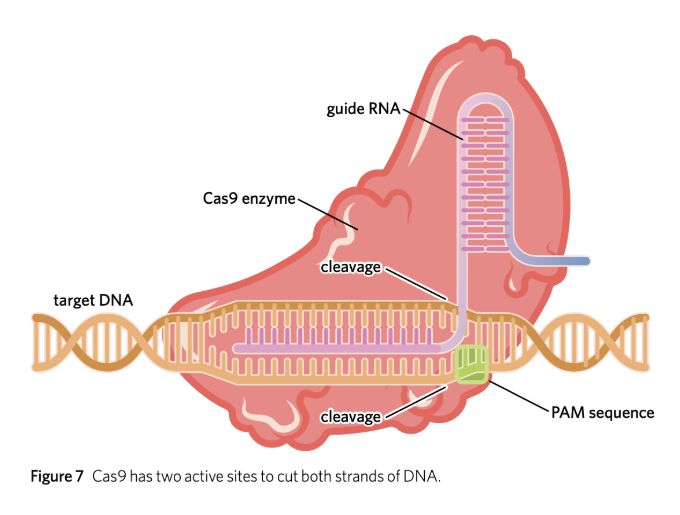
CRISPR-CAS9
a complex formed between gRNA and Cas9 which can cut a target sequence of DNA
Bacteria uses this complex for protection from viruses
Scientists modify this complex to edit genomes
Cas9
an endonuclease that can be programmed to cut any specific DNA recognition sites
sgRNA (single-guide RNA)
guides Cas9 and tells it where to cut.
Made up of ribose sugar, phosphate and nitrogenous bases
Changes in sgRNA determines how Cas9 is programmed
Scientists can create sgRNA with any sequence to cut at any known target gene
Effect of temperature on Cas9 reaction rate
Reaction rate is greatest at optimum temperature
Above opt. temp. → enzyme denatures → decreasing rate of reaction
Below opt. temp. → less kinetic energy (fewer collisions) → slow rate of reaction
FIRST step of CRISPR-Cas9 in bacteria
Exposure
Bacteriophage (virus) injects its DNA into a bacterium + recognises the viral DNA as foreign
Cas1 and Cas2 cut a short section of viral DNA (protospacer) as it matches the PAM
Stored in the bacterium’s CRISPR array as a spacer (+ bacterium now has a genetic “memory” of the viral DNA)
SECOND step of CRISPR-Cas9 in bacteria
Expression
When virus attacks again, CRISPR array is transcribed into guide RNA (gRNA)
gRNA binds to Cas9 to create a CRISPR-Cas9 complex
It is directed to the viral DNA complementary to gRNA sequence
THIRD step of CRISPR-Cas9 in bacteria
Extermination
CRISPR-Cas9 complex scans the call for viral DNA complementary to gRNA sequence
Upon finding target, Cas9 cleaves viral DNA by making a double-strand break in the sugar-phosphate backbone to inactivate the virus
After successful cleavage, new viral sequences are added to CRISPR array to protect bacterium from future attacks by the same virus
CRISPR-Cas9 in Gene Editing — Process
Identify the sequence of the gene to be targets
Create sgRNA complementary to the target gene
Combine sgRNA with Cas9 and introduce into organism
sgRNA + Cas9 looks for PAM sequence on DNA strand
When PAM sequence is found, Cas9 looks for target gene sequence
DNA is unwinded to let Cas9 cut the target gene
DNA sequence is altered (by additional processes) to either switch off or repair/alter the gene
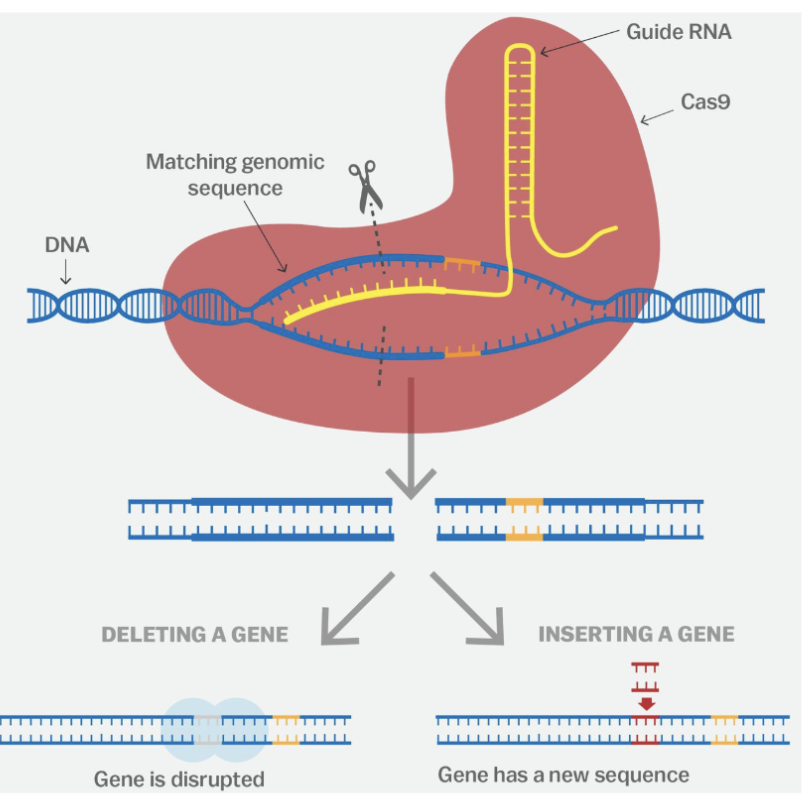
Gene Knocks IN Gene Editing
a new DNA sequence is inserted into the DNA break
Allows a faulty gene sequence to be replaced with the correct one to restore gene function
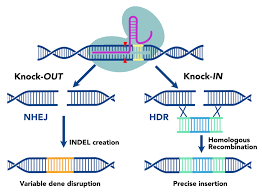
Gene Knocks OUT Gene Silencing
errors (mutations) can occur as the cell’s normal repair mechanisms mend the broken DNA, causing the insertion or deletion of bases
Changes the way the nucleotide sequence is read
Disables gene function + produces a STOP signal
SILENCES the gene
PAM (Protospacer Adjacent Motif)
a very short segment (2-6) of nucleotides (NGG) on the DNA that provides a binding site for Cas9
Without PAM, Cas9 will not cut DNA even if a sequence complementary to the gRNA is present
Makes CRISPR more efficient
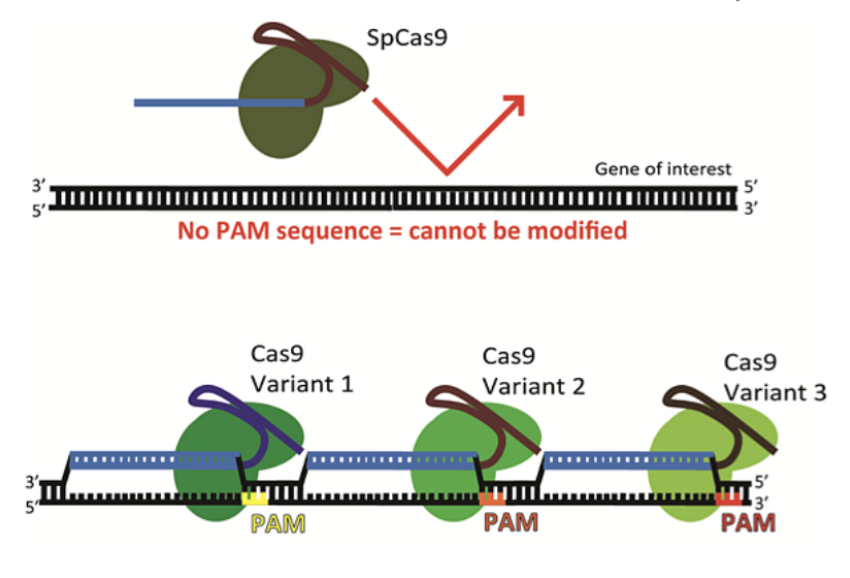
Role of PAM
Protective of bacteria
Bacteria never have a PAM sequence in their own DNA
- Ensures Cas9 cannot cut the bacterium’s own DNA
PCR (Polymerase Chain Reaction)
PCR amplifies a specific DNA sequence by making multiple identical copies
Used by scientists when there is insufficient DNA samples for testing
Entire genomes are not copied, but certain genes are, to make process more efficient
After each PCR cycle, amount of DNA is DOUBLED
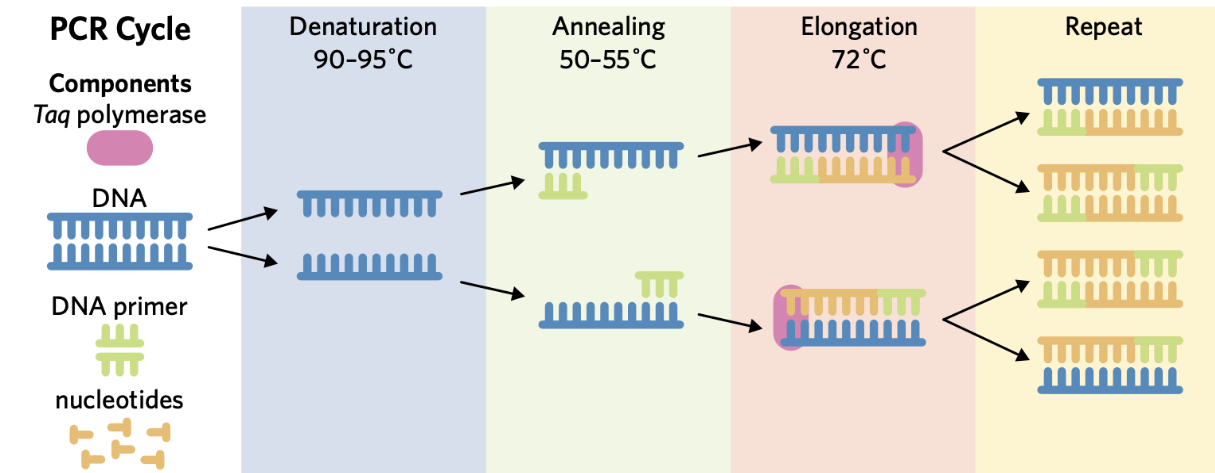
Purpose of PCR
amplifies a sample of DNA to increase the quantity of DNA available
FIRST step of PCR
Denaturation
DNA is heated to ~90-95℃ to break the hydrogen bonds between the bases and separate the strands, forming single-stranded DNA
SECOND step of PCR
Annealing
The single-stranded DNA is cooled to ~50-55℃ to allow the primer to bind to complementary sequences on the single-stranded DNA
THIRD step of PCR
Elongation
The DNA is heated again to ~72℃, the optimal temperature for DNA polymerase to synthesise new DNA strands by adding nucleotides in 5’ to 3’ direction
FOURTH step of PCR
Repeat
The cycles (steps 1-3) is repeated multiple times to create more copies of DNA
Forward Primers
binds to the start codon at the 3’ end of the template strand
Causes Taq polymerase to synthesise a new DNA strand in the same direction that RNA polymerase would function
Reverse Primers
binds to the stop codon at the 3’ end of the coding strand
Causes Taq polymerase to synthesise a new DNA strand in the reverse direction that RNA polymerase would function
Importance of Primers
Having two primers is necessary as the 5’ ends of both the template + coding strands are different
As Taq polymerase only functions towards the 3’ end, a primer is needed for both strands to facilitate directionality
Taq polymerase vs. DNA polymerase
Taq → very high optimal temperature, working optimally at 72℃
DNA → would denature, being incapable of synthesising a new strand at that temperature
Gel Electrophoresis
a technique used to compare DNA samples by separating DNA fragments according to their size
DNA samples are often amplified using PCR + cut using restriction enzymes BEFORE being separated
Gel is made of agarose, immersed in a buffer solution
DNA is stained with ethidium bromide to see more clearly under UV light
An electric current is used to separate the fragments
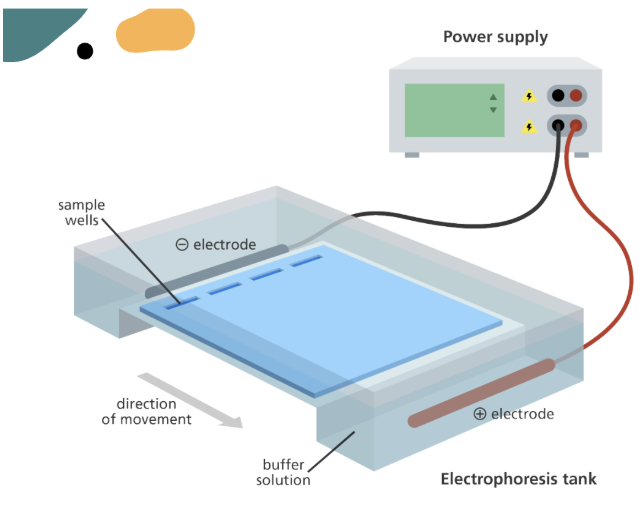
Wells
DNA samples are placed in wells in the gel
These wells are always placed at the negative electrode as DNA has a negative charge
Usually the first well contains a sample with DNA fragments of a known size (DNA ladder)
Gel Electrophoresis — Process
When the power is turned ON → the DNA moves through the gel towards the positive electrode
Because DNA has an overall negative charge
Smaller DNA fragments move more quickly + travel further through the gel
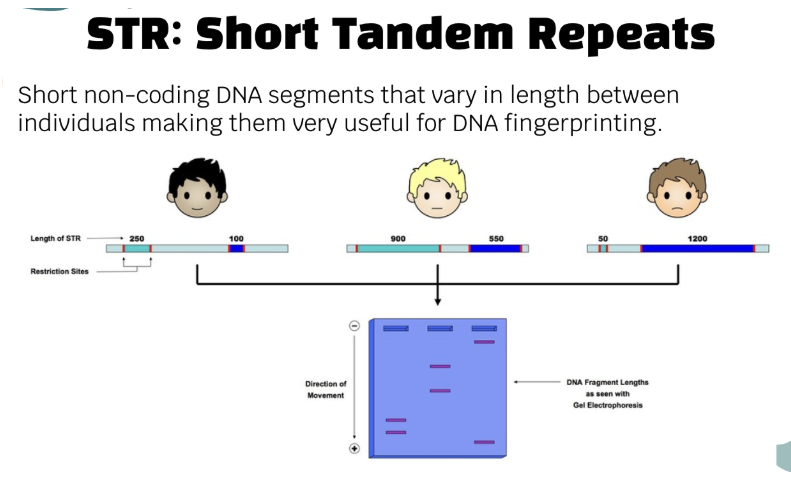
STR (Short Tandem Repeats)
short non-coding DNA segments that vary in length between individuals making them very useful for DNA fingerprinting
Bacterial Transformation
when scientists insert new genes into bacteria + turn them into a protein-producing factories
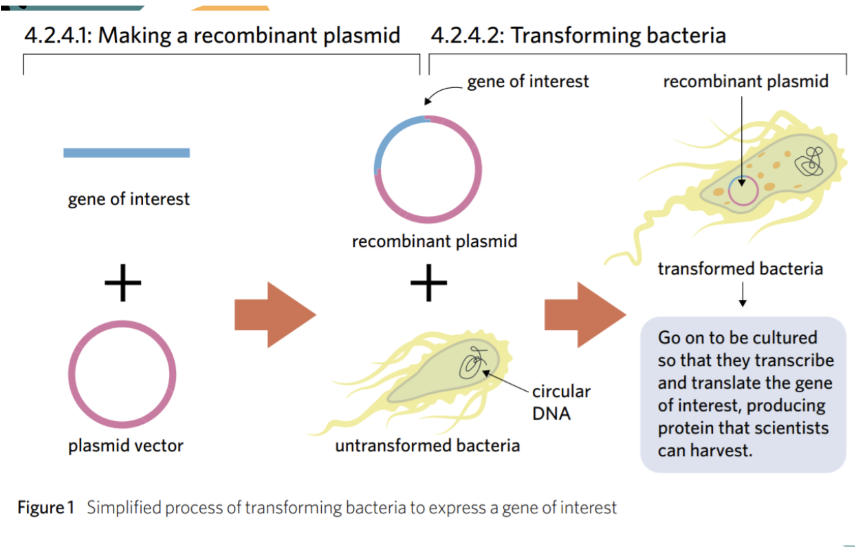
gene of interest
the gene we want the bacteria to express (e.g insulin gene)
plasmid vector
a circular ring of DNA used to transport the G.O.I into the bacteria
Recombinant plasmid
plasmid that has the G.O.I inserted into it (basically G.O.I + P.V)
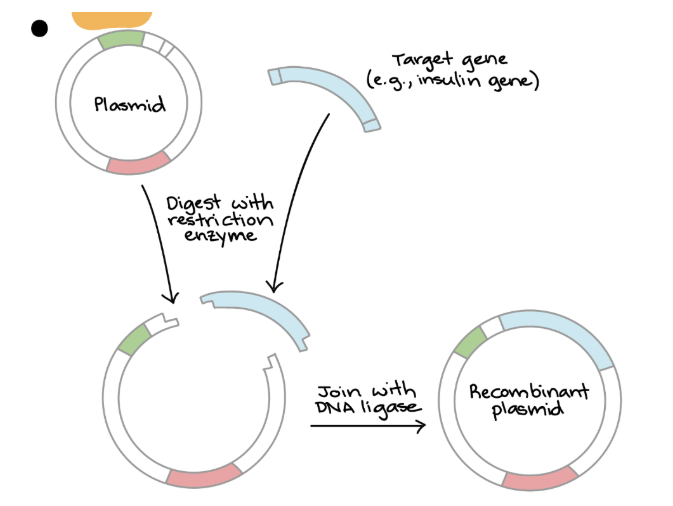
Restriction endonucleases
cuts G.O.I + P.V, producing complementary “sticky ends” that allow the gene to be inserted into the plasmid
DNA ligase
joins G.O.I + P.V together by forming phosphodiester bonds between each DNA sugar-phosphate backbone
Creating a Recombinant Plasmid — Process
1. The gene of interest is removed from the DNA strand using restriction enzymes
2. The plasmid is also cut using the SAME restriction enzyme
3. G.O.I + plasmid have complementary sticky ends + easier to join
4. G.O.I is positioned in plasmid → ligase joins the two pieces of DNA
(Steps 1-3) Bacterial Transformation — Process
1. The recombinant plasmid combines with bacterial cells
2. Mixture is treated with heat/electricity shock to encourage bacteria to take up the recombinant plasmid
3. Bacteria that take up the recombinant plasmid are transformed
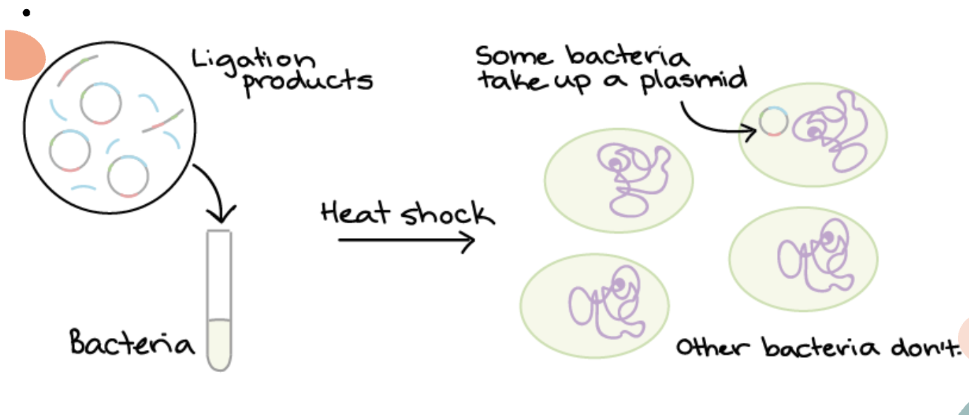
FOURTH step of Bacterial Transformation — Process
4. Not all bacteria will take up recombinant plasmid
Only the bacteria with antibiotic resistance gene (transformed) will survive
Allows us to only culture the transformed bacteria that will express G.O.I
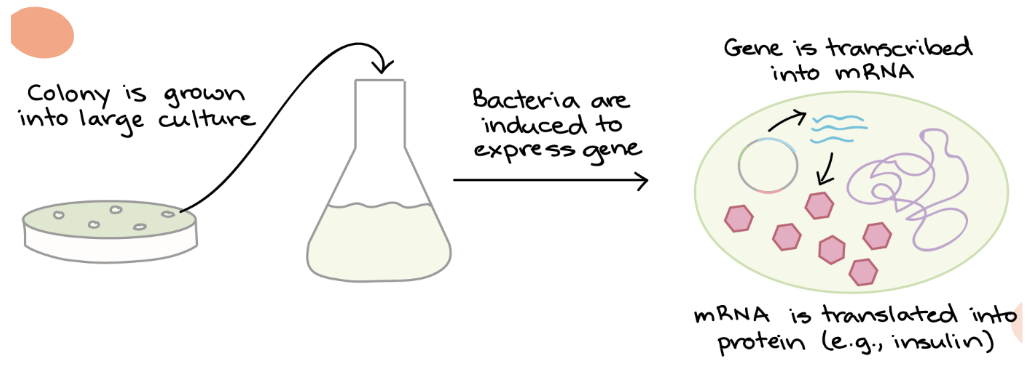
FIFTH step of Bacterial Transformation — Process
5. Colonies of transformed bacteria should be observed on nutrient agar
Transformed → should form colonies as they contain tcl gene, conferring resistance to tetracycline in the culture
Untransformed → unable to form colonies as they do not contain tcl gene and die
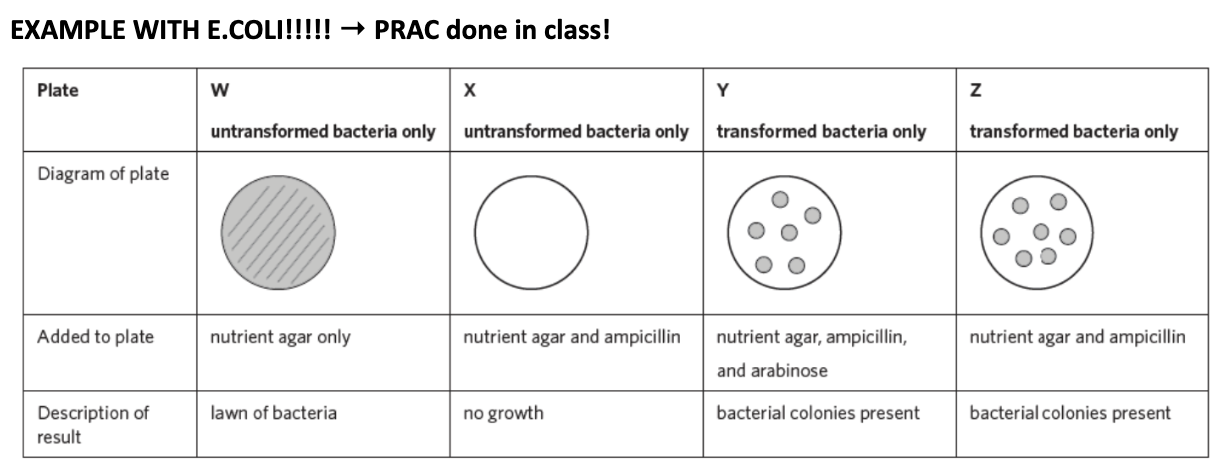
EXAMPLE WITH E.COLI → PRAC DONE IN CLASS!
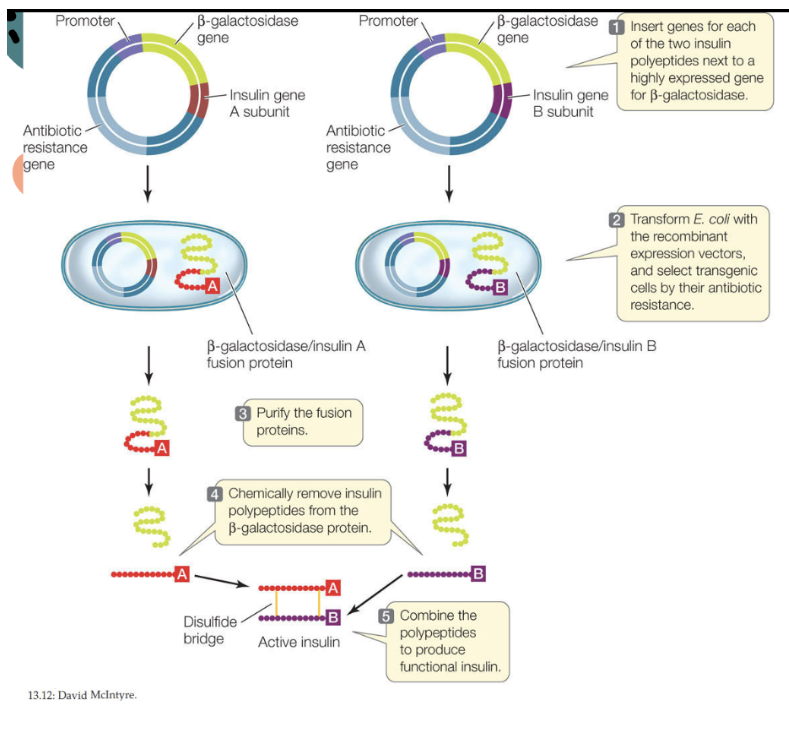
Human Insulin
has a quaternary structure, made of 2 polypeptide chains
Two separate plasmids must be produced + placed in separate bacteria → one for Chain A and Chain B
When the polypeptides are extracted, they are combined to form functional human insulin
Reporter Genes
used in bacterial transformation to indicate whether the G.O.I is being expressed or not
Code for proteins that are easily visualised
Allows researchers to select for bacteria that have taken up the recombinant plasmid (will express G.O.I) → similar to antibiotic resistance gene (with/without G.O.I)
Examples of Reporter Genes
GFP → causes bacteria to glow green
Lac z → causes bacteria to turn blue
GMO — Genetically Modified Organism
an organism that has had its DNA artificially altered in any way.
Examples of GMOs
Inserting DNA from SAME species
Removing DNA
Silencing genes
Replacing nucleotides
TGO — Transgenic Organism
an organism that has had a gene from a different species inserted into its genome
Example of TGO
Inserting DNA from ANOTHER species
Non-GMO
an organism that genetic material has not been altered using genetic engineering or other modern biotechnology techniques
Examples of Non-GMO
Selective Breeding
Vaccination