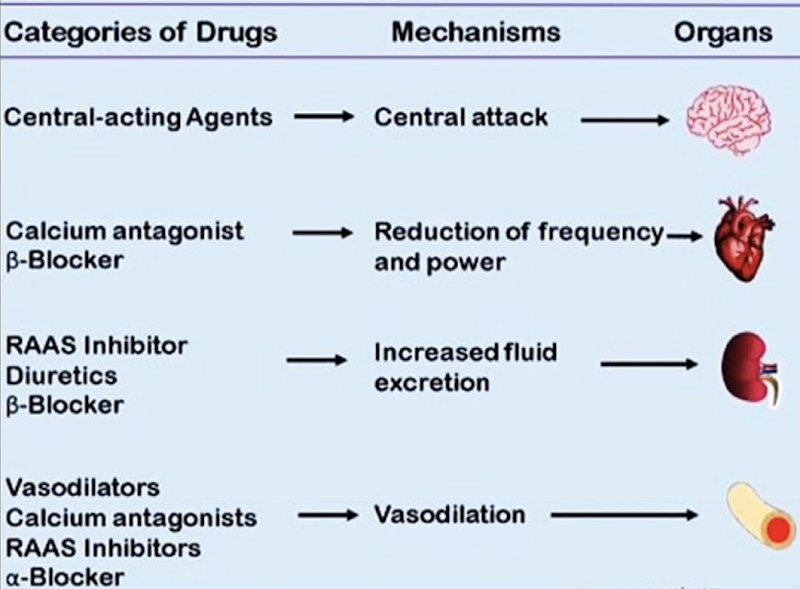
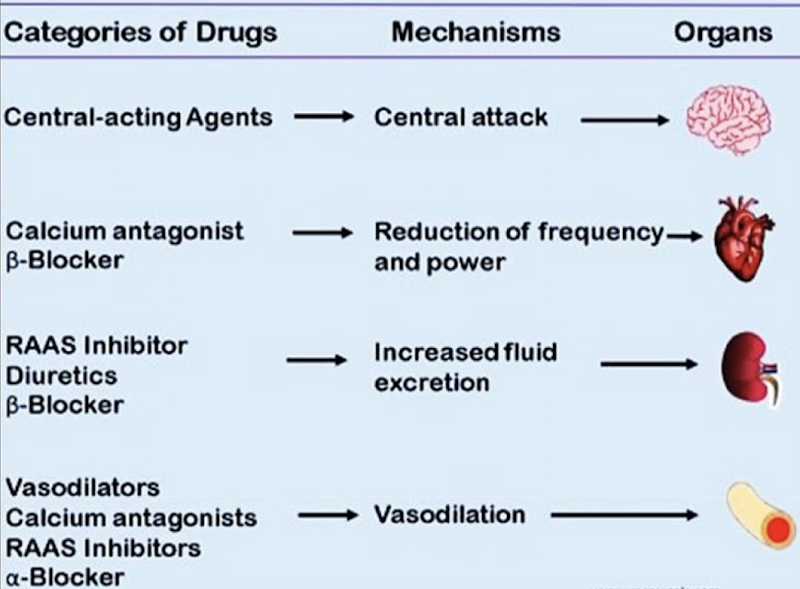
PHAR 300 midterm 1 notes
1/135
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
136 Terms
Chemical name
Name that is representative of the chemical structure of the molecule.
Ex.: 2-(acetyloxy) benzoic acid is the chemical name for Aspirin.
Generic name (nonproprietary)
Name that we will use in class!
Universal name assigned by the United States Adopted Name Council (USAN) and by WHO International Nonproprietary Names program (INN)
Purpose: to create a unique, universal name that indicates what the drug does
Structure: contains a prefix (specifies what the drug does), followed by a stem (identifies the class of the drug, provides some information on the drug’s action)
Ex.: Trastuzumab (anticancer drug)
Prefix: -tu indicates that the drug targets a tumor
Stem: -zumab indicates that the drug is a humanized monoclonal antibody
Trade name (proprietary)
A registered trademark (different for each manufacturer; use restricted to owner)
There can be multiple different trade names for a drug. They are often catchy and easy to remember
Ex.: Ciprofloxacin (generic) is also known as Cipro, Ciloxan, Neofloxin, etc. (trade)
Pharmacodynamics
What the drug does to the body. Observes the biological action of a drug on the target cell (through interactions with receptors, ion channels, enzymes, and the immune system) or the organ system.
Pharmacokinetics
What the body does to the drug. Describes what happens to the drug after its ingestion. Absorption, Distribution, Excretion, Metabolism.
Selective drugs
Highly selective drugs act on a very specific drug target → prevents drug activity (side effects).
Ex.: Antiviral drugs target a feature of the virus that is not present in human cells. In most cases, the antiviral drug acts only on the virus (by binding to viral proteins, for example), and results in few side effects for the patient.
Generalized drugs
Drugs with a generalized effect act on targets that are present throughout the body.
Ex.: Analgesic drugs (for pain relief), like opioids, have multiple drug targets because there are opioid receptors everywhere in the body. While this drug does manage to relieve pain, it also causes side effects due to its ability to act on a wide variety of cells.
Ex.: Intravenous administration of epinephrine causes a generalized effect. It is used in cases of severe allergic reactions
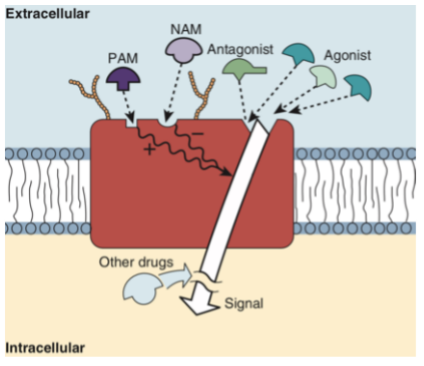
Receptors
A macromolecular protein target to which endogenous ligand or exogenous agonist/antagonist binds to cause a cellular response.
Criteria for receptors
The receptor may be located on or in the cell.
Respond to neurotransmitters, hormones, growth factors, cytokines, etc.
Must be linked to cellular response elements (ex. Ion channels, enzymes, second messengers, etc.)
Receptors are identified by radioligand binding. These ligands are then isolated and sequenced, allowing us to understand their structure. This is an important step in drug design.
Drugs can bind to the receptor (allosterically or to the active site), or act on other substances in the signaling pathway to elicit a change.
Forms of chemical signaling
Autocrine: cell secretes a chemical to act on its own surface receptors
Juxtacrine: cell secretes a chemical that acts on a neighbor cell via a connection
Paracrine: cell secretes a chemical that acts on a nearby cell
Endocrine: cell secretes a chemical that acts on a different cell after being transported through the blood
Types of receptors?
Intracellular receptors
Receptor-activated enzymes
Receptor-activated tyrosine kinase
Receptor-activated ion channel
G protein-coupled receptors (GPCR)
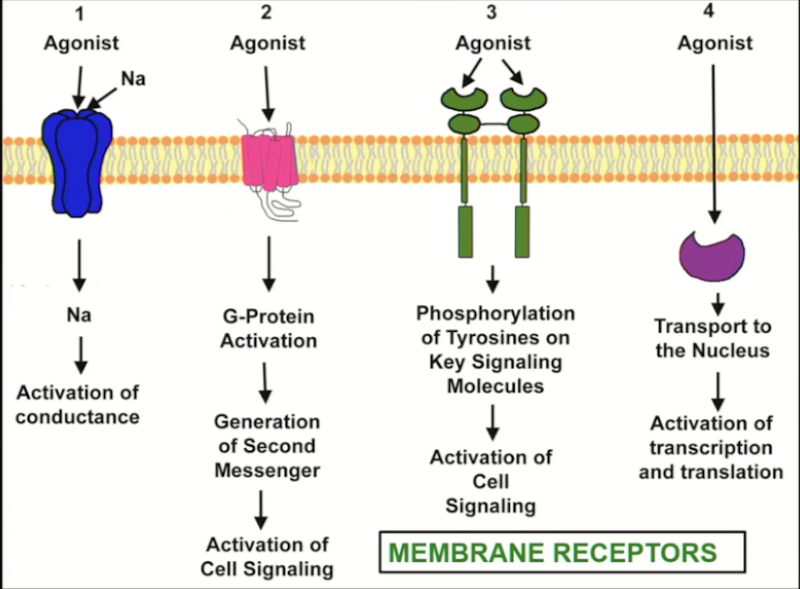
Response time of receptors
Ion channel: fast to respond (milliseconds)
GPCR: second messenger system must be activated (seconds)
Enzymes: blocking or stimulating enzymes (minutes)
DNA-linked: translocation, transcription, translation, enzymatic activity (hours)
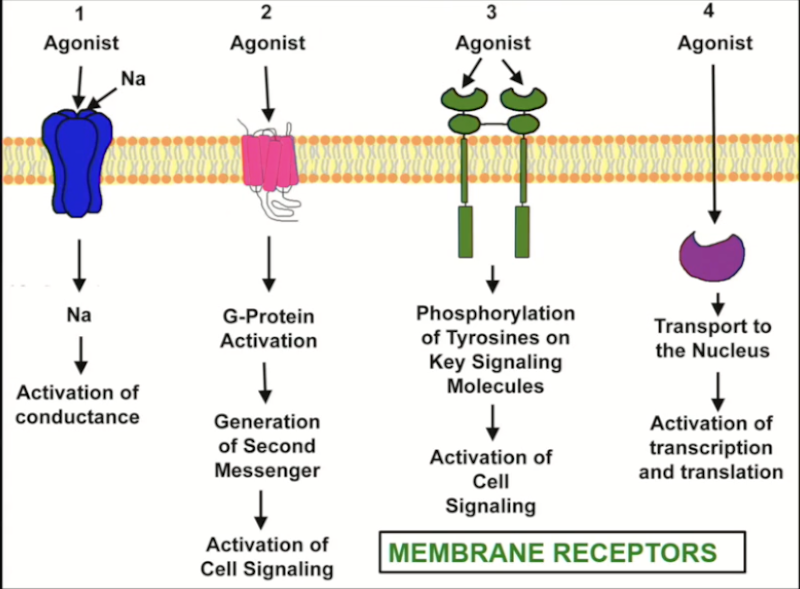
Ion channels
Transmembrane-spanning proteins that open to allow passage of specific ions.
Found all over the body
Present in 3 conformations: open, closed, inactivated
Can be voltage or receptor (ligand) controlled
Drugs can both activate or block ion channels.
G-protein coupled receptors
Biggest category of receptors found in the body.
Transmembrane receptor spanning the plasma membrane 7 times, coupled to a G-protein (3 subunits: α, β, γ)
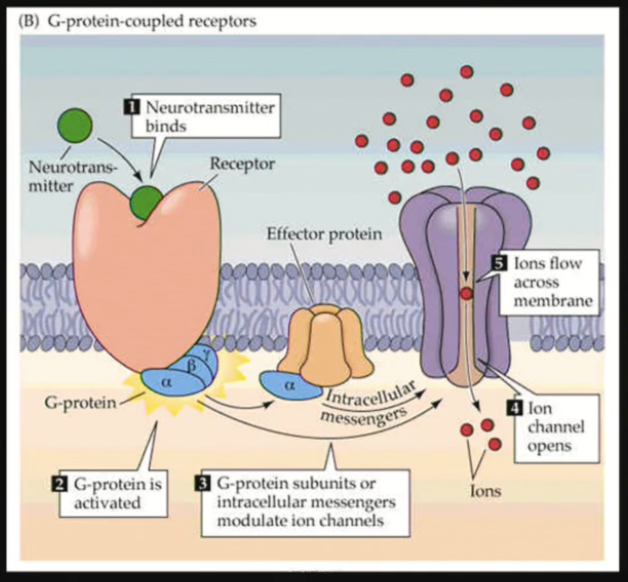
GPCR activity
Ligand binding
Activation of the G-Protein: GDP bound to the α subunit is exchanged for GTP (ON state)
Activation of an effector by the active α subunit (ex.: enzyme, ion channel)
Changes inside the cell/downstream signaling
Bound GTP is exchanged for a GDP = Inactivation of the α subunit
Multiple receptors modulate the same pathway: GPCRs may have opposite effects (stimulatory and inhibitory) on a system. Balance between the stimulatory and inhibitory signaling regulates the effect.
May form dimers or oligomers (homo- or hetero-), which alters the downstream effects.

Enzyme-linked receptors
Receptors in this class must dimerize in order to signal.
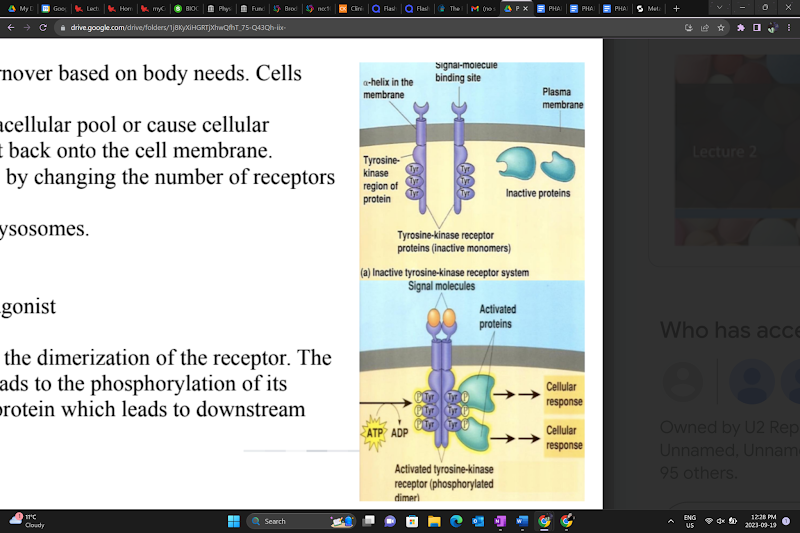
Receptor Tyrosine Kinases (RTKs)
The ligand binding to the receptor leads to the dimerization of the receptor. The tyrosine kinase receptor is activated and leads to the phosphorylation of its tyrosine residues. This further activates a protein, leading to downstream signaling.

Cytokine receptors
Receptor activation → dimer → phosphorylation of Janus kinase and STAT (signal transducers and activators of transcription) proteins on tyrosine residues
STAT proteins translocate to the nucleus, activating transcription
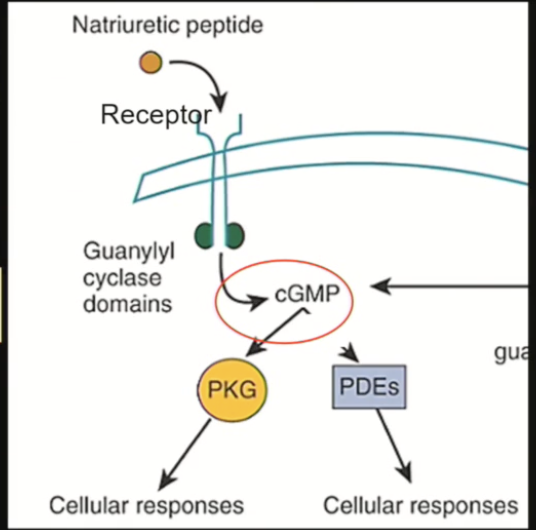
Natriuretic peptide receptors (NPRs)
Hormone binds, receptor dimerizes, activates enzyme to produce cGMP, which controls many cellular responses
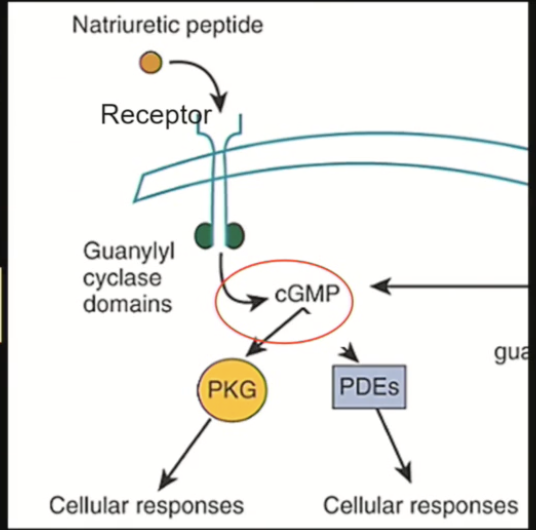
Receptor turnover
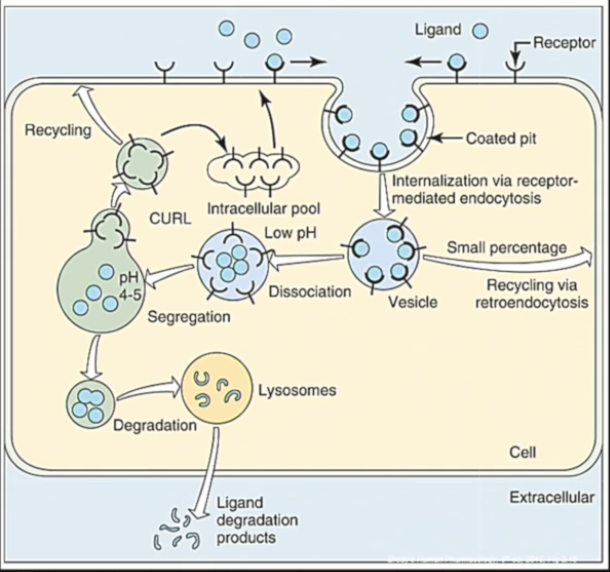
Receptors are dynamic: there is a constant turnover (synthesis, transport, stimulation, recycling, resynthesis etc…) based on the body’s needs.
Receptors can be internalized to enter an intracellular pool or cause cellular signaling and can be further recycled and put back onto the cell membrane.
The action of a drug on a cell is terminated by the internalization of its receptor, and its degradation.
With chronic drug use, the number of receptors changes to counteract the effects of the drug.
Intracellular receptors
Hormone receptors
Steroid/steroid receptor complex translocates to the nucleus, binding to DNA and triggering or blocking transcription → alteration of gene transcription and protein synthesis
Receptor has a ligand binding domain (LBD) and a DNA binding domain (DBD)
Ligand – LBD binding activates the receptor, which allows for DNA – DBD binding

Enzymes
Enzymes can alter synthesis or breakdown of transmitters, cytokines, hormones, etc.
Drugs can bind to an enzyme and stimulate/block it → change in synthesis or breakdown of other molecules
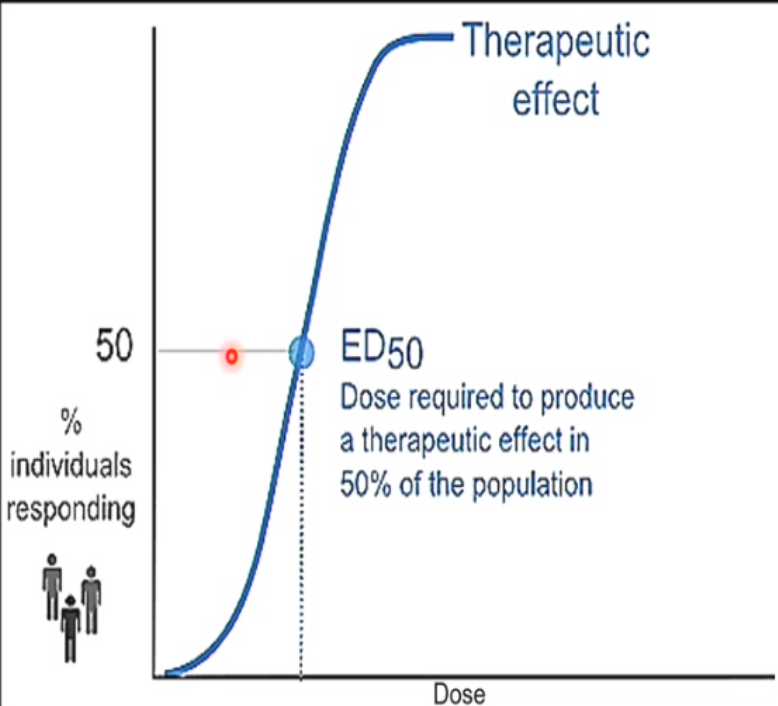
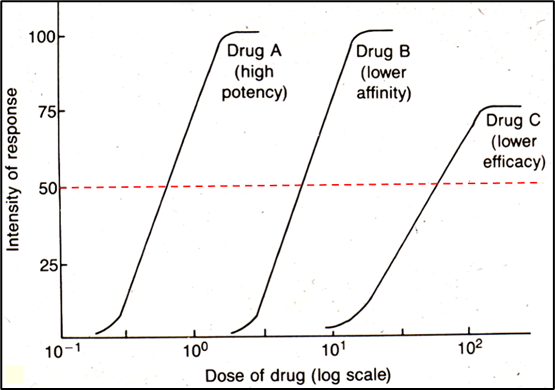
Dose-response curve
Curve used to show the relationship between the dose of a drug (x-axis, log scale) and the percentage of individuals experiencing a therapeutic effect (y-axis).
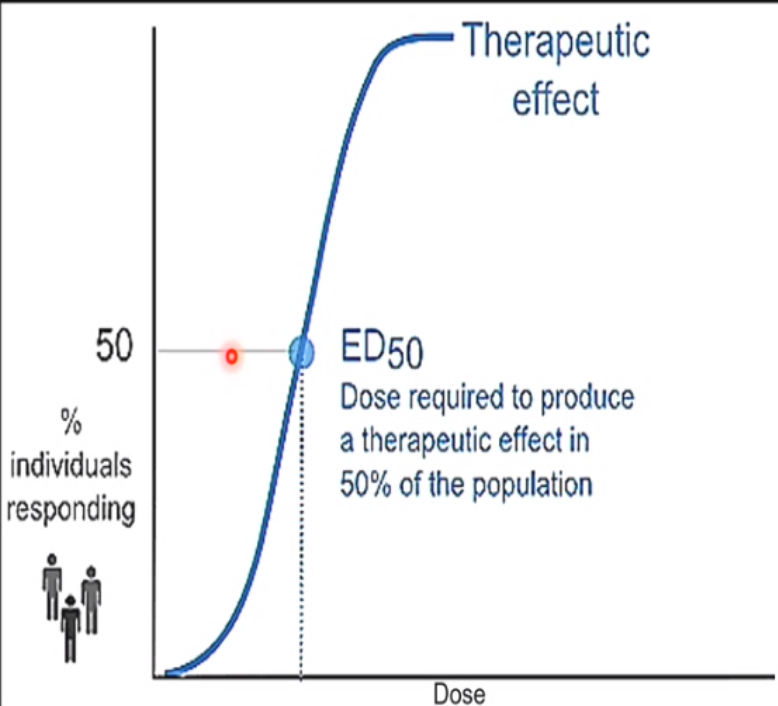
ED-50 (effective dose - 50)
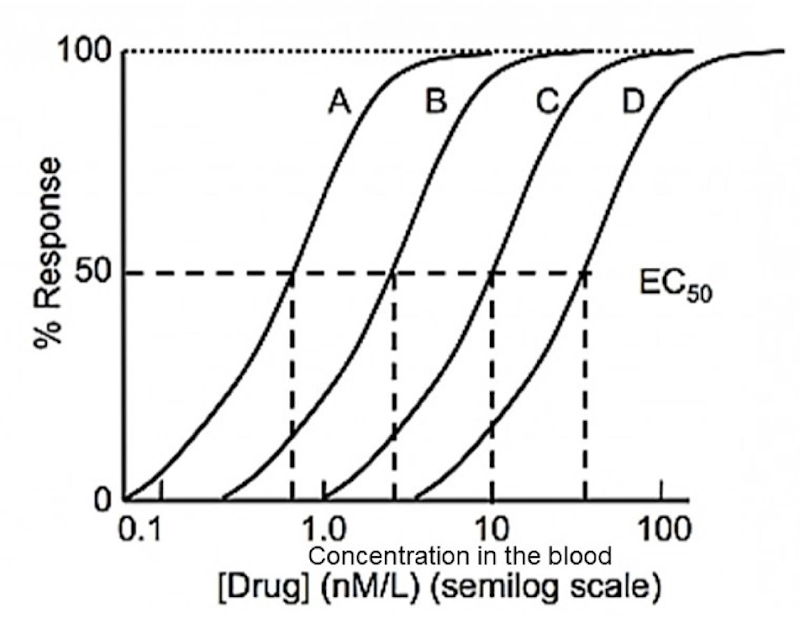
Which drug is the most potent
Drug A
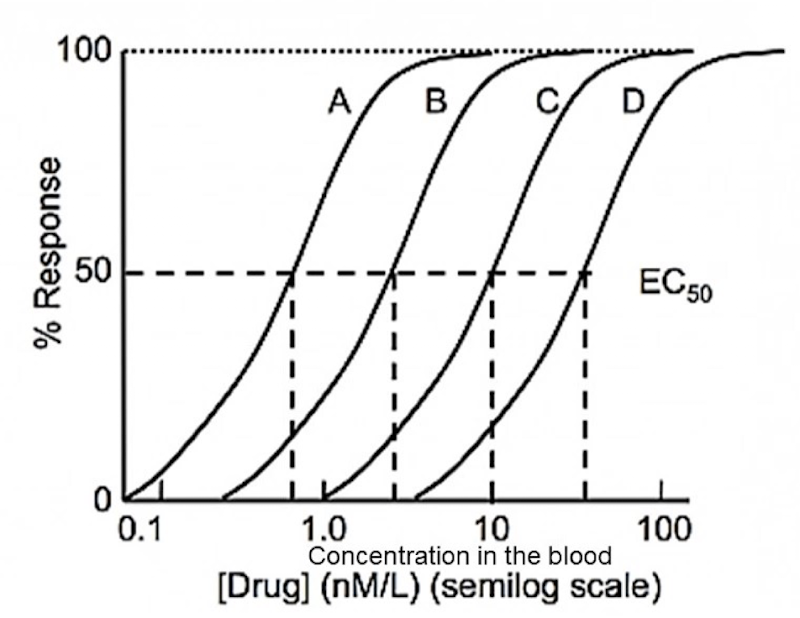
Would be ALWAYS want to use drug A?
Not necessarily – Both the therapeutic and toxic effects of a drug must be taken into account to make an informed decision. This graph omits the toxicity of these drugs (drug D may end up being much safer than Drug A).
Explain a situation where the dose response curve shifts left
Ex.: Chronic administration of an antidepressant treatment on a neuron leads to an increase in serotonin (5-HT) receptor sensitivity over time – supersensitive response.
Explains why a dose-response curve can shift (in this case, to the left)
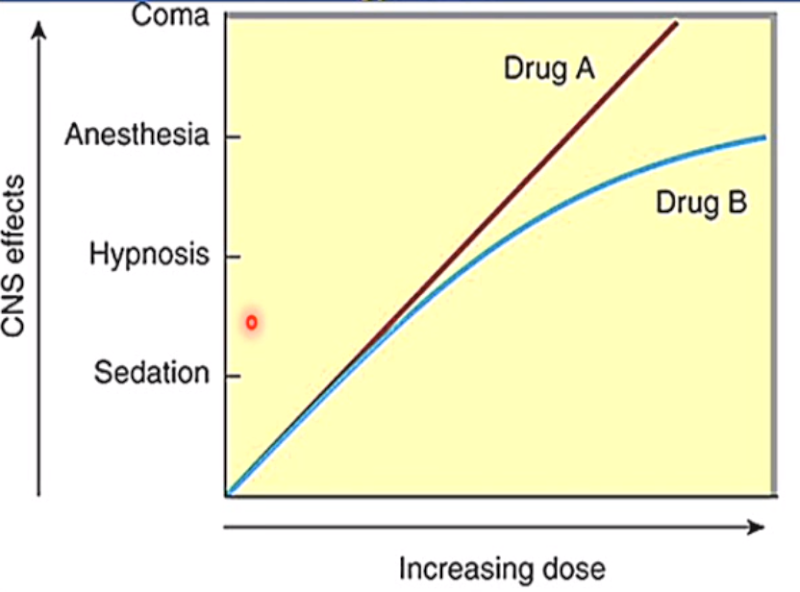
Which drug is safer?
Ex.: Drug B is safer than Drug A because it plateaus. Drug A continues to depress the CNS beyond the wanted therapeutic effects. Excessive CNS depression could be lethal.
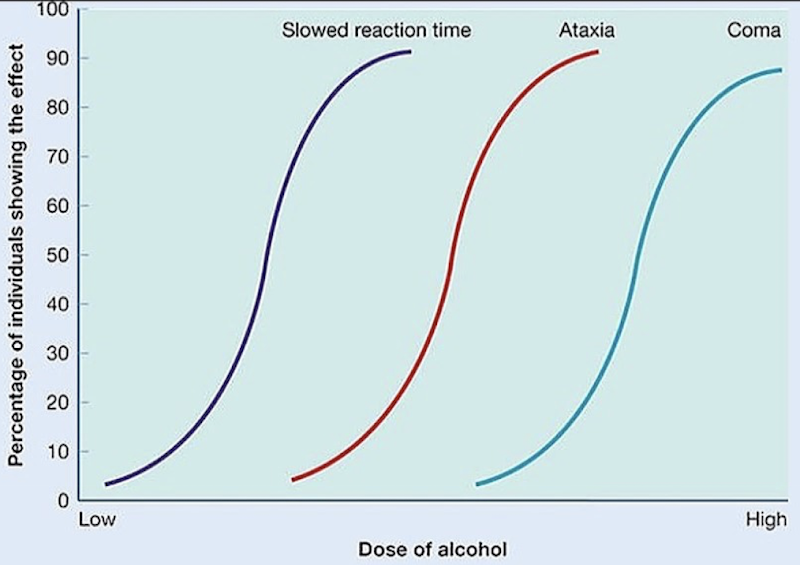
How can different doses of alcohol impair the CNS?
Shows how different doses cause different effects across a population, helping compare therapeutic vs. toxic effects
Ex.: Progressive CNS impairment with increasing alcohol dose; Slowed reaction time → ataxia → coma. Shows that reaction time is affected early on compared to more toxic effects of alcohol.
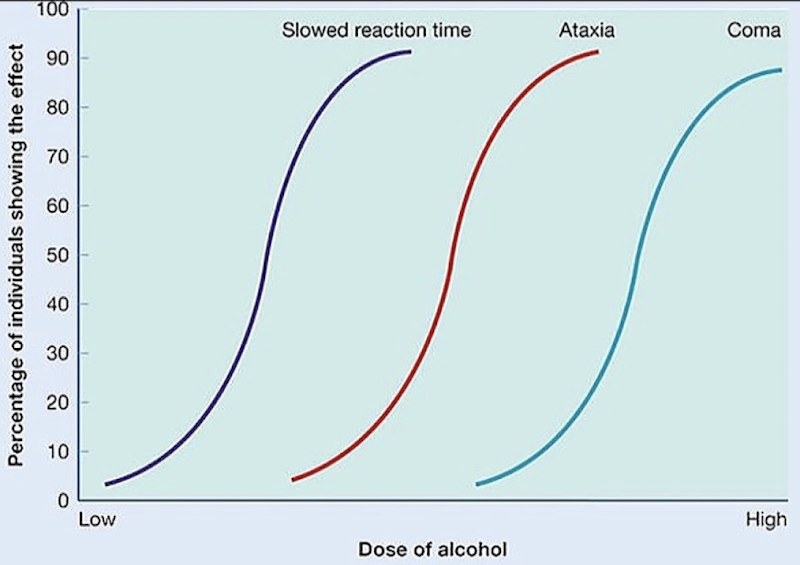
Dose-response curves (not a question)
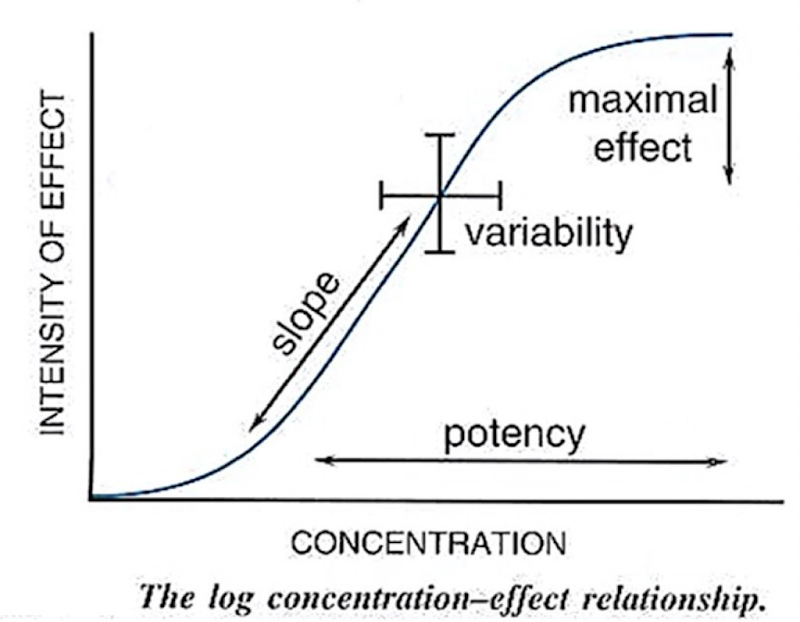
Threshold: The lowest dose/concentration at which a measurable effect is observed (where the curve begins to rise)
Ceiling dose (maximal effect): The dose at which the maximal effect of the drug is observed; beyond this dose, increasing concentration does not increase effect.
Potency: The dose required to produce an effect of a given intensity. Comparative measure between drugs, not a fixed value
Slope: indicates how rapidly effect changes with concentration.
Variability: reflects differences in response among individuals.
How does a high and low potency drug shift the dose response curve?
High potency: curve further to the left, since a smaller dose can produce a therapeutic effect
Low potency: curve further to the right, since a larger dose is needed to produce a therapeutic effect
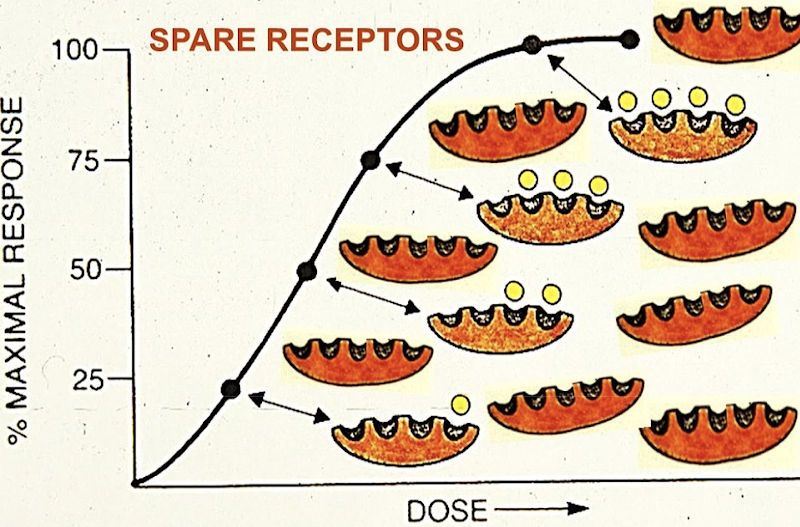
Do all the receptors need to be bound to elicit a response?
No
How does affinity and efficacy effect a drug?
Drug affinity influences drug potency.
Efficacy affects the height of the dose-response curve. Low efficacy will result in a lower maximal response.
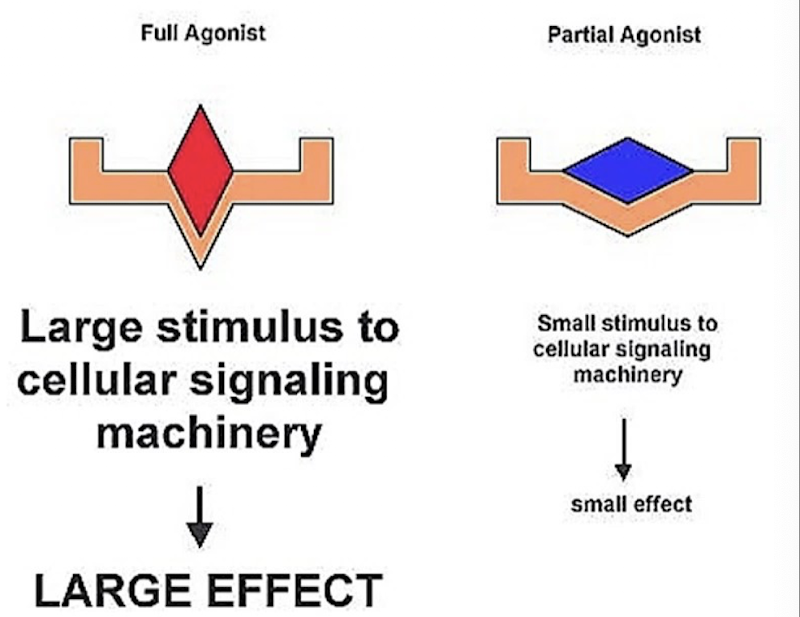
Partial vs full agonists
Full agonists fully activate the receptor, leading to a large cellular response → High efficacy
Partial agonists bind to the receptor, but result in a smaller cellular response → Low efficacy
Partial agonists can lead to less adverse effects
Even with high receptor occupancy, the physiological response to a partial agonist may be low.
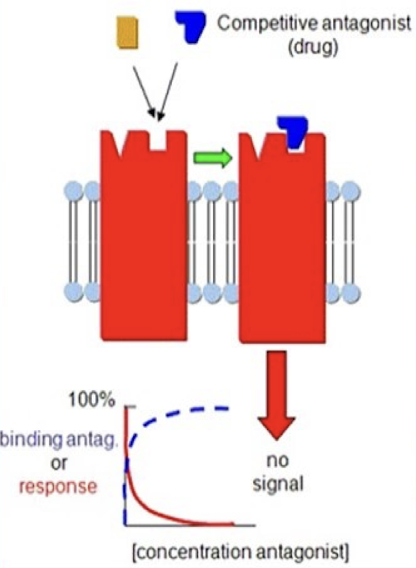
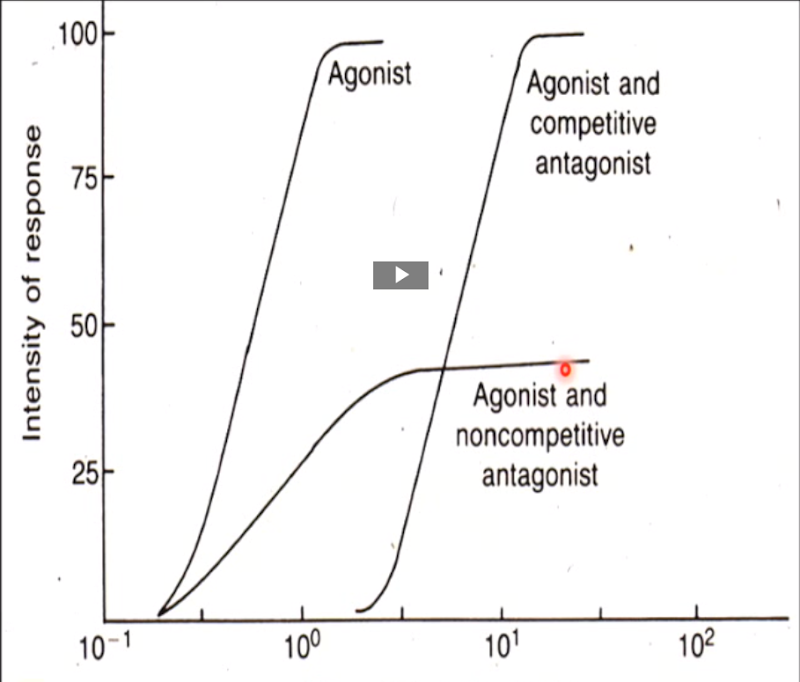
Competitive antagonists
Bind to the same site as the agonists to block the agonist’s effect.
Their blockade can be overcome by increasing the agonist concentration, allowing a full response (100%) to be reached, though at higher doses than normal due to receptor competition. Spare receptors make this possible, since enough receptors can still be activated despite competition.
At very high doses, a competitive antagonist can occupy so many receptors that the agonist can no longer achieve its maximal response.
Shift the dose-response curve to the right
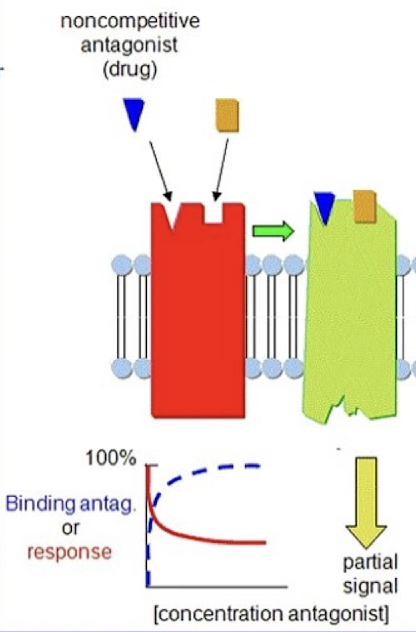
Non-competitive antagonists
Bind to a different site than the agonist to block its effect.
Modulate the effect of agonists, either reducing it or entirely blocking it.
Allosteric: antagonists that bind to an allosteric site on the receptor → change the conformation at the active site to prevent agonist binding.
Intracellular: agonists that act inside the cell on parts of the signaling machinery and prevent its normal functioning.
The effect of the non-competitive antagonist cannot be overcome with an increased dose of agonist.
Lowers the peak of the dose response curve (lower maximal response)
Irreversible antagonists
Bind to the receptors permanently and irreversibly → longlasting effects
Decrease the number of available receptors, forcing cells to increase the receptor turnover rate
Reversible antagonists
Bind to the receptors reversibly → short duration of effects
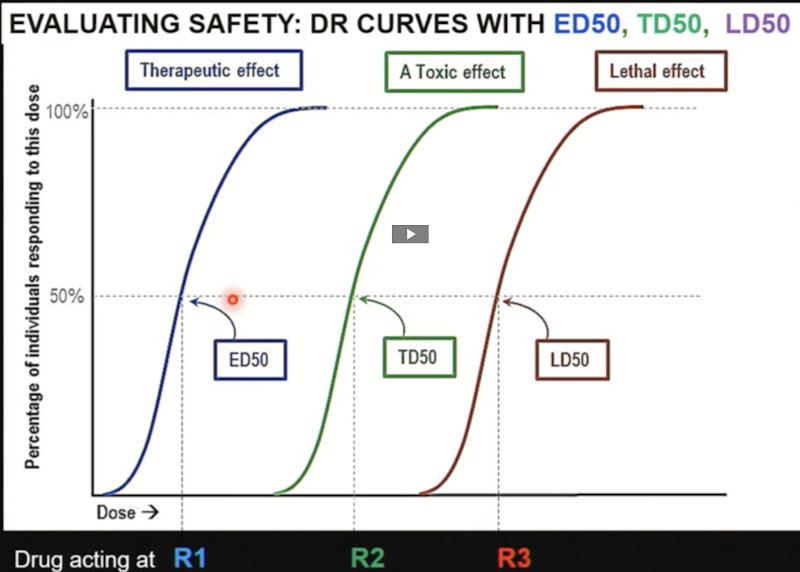
ED50 vs TD50 vs LD50
● ED50: The dose of drug required to produce a therapeutic effect in 50% of the population.
● TD50: The dose of drug required to produce a toxic effect in 50% of the population.
o Toxic effects are non-lethal side effects (nausea, headache, diarrhea, etc.)
● LD50: The dose of drug required to produce a lethal effect in 50% of the population
Therapeutic window
The dose range within which most people get therapeutic effects without side effects.
The ideal is to have the toxicity curve as far away from the therapeutic curve as possible. (bigger window)
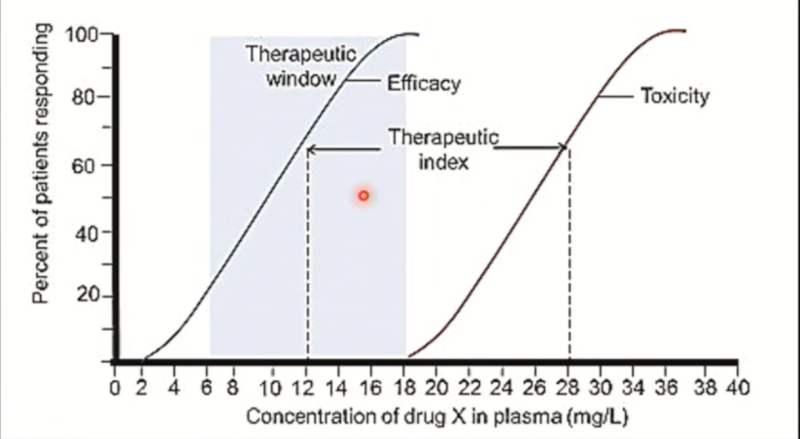
Therapeutic window: Penicillin vs. Warfarin
100% of people will achieve the desired effect of penicillin at doses that are WAY below the unpleasant side effects (large therapeutic window). Penicillin is remarkably safe, as it is highly selective for microbial substances not present in humans.
Warfarin, on the other hand, has a small therapeutic window. The dose required to achieve a therapeutic effect in everyone often overlaps with the unwanted side effects.
Drugs with a small window are used when there are no other alternatives, like Warfarin here, an anticoagulant, in cases of risk of blood clots.
Therapeutic index (TI)
The ratio of toxic effect to the therapeutic effect:
The larger the therapeutic index, the safer it is.
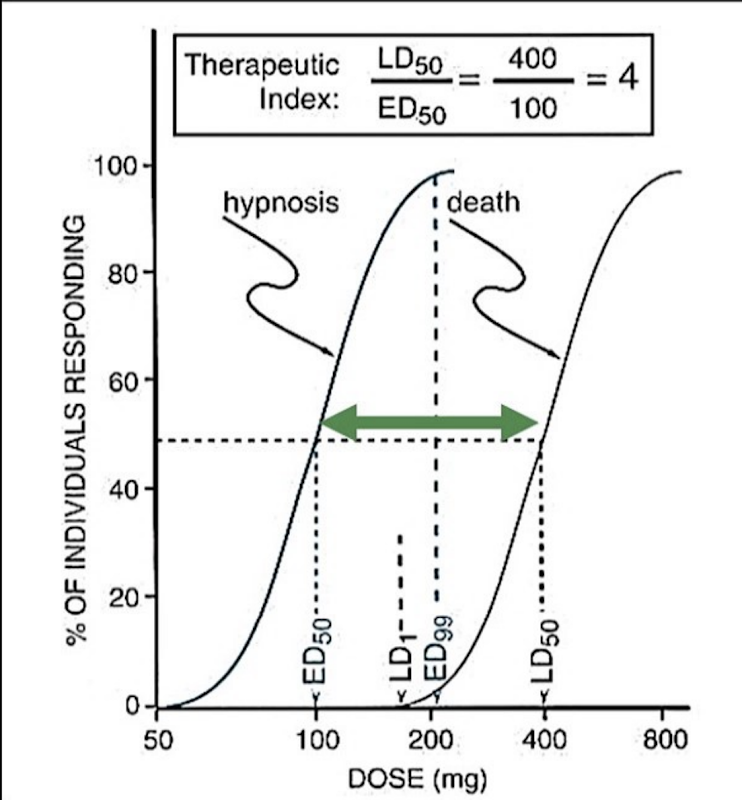
Safety factor
TD1/ED99:
The larger the safety factor, the safer the drug.
The dose that causes toxicity in 1% of the population VS the dose that's effective in 99% of the population.
Can we rely on the therapeutic index if the slopes of the toxic and therapeutic curves are different?
No, you must look at the entire dose-response curve:
Both drugs (blue and yellow) have the same ED50 and TD50
Blue has no overlap = safe
Yellow has an overlap = not safe because the dose that you need to use to be effective in most people is going to cause side effects in some people.
Thus, you need to know more than just the ratio because TI does not tell us everything about the slope!

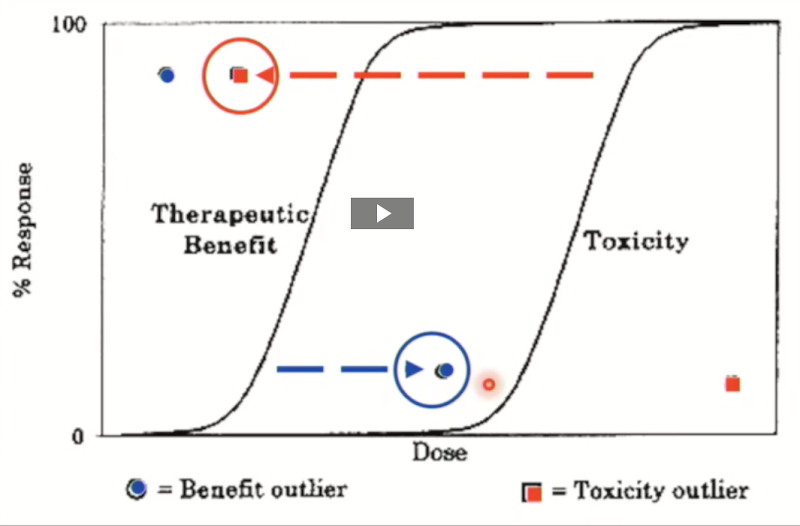
Benefit vs toxicity outlier
o Benefit outliers (Not a problem)
Drug works at a very low dose
Little/no toxic effect even at high dose
o Toxicity outliers (ineffective or toxic)
Shows serious toxicity at very low dose
Need very high doses to see an effect → toxicity risk.
Oral administration
Easiest, cheap, convenient:
o Modified released tablets exist
o Oral drugs must be absorbed through the GI tract
o Involves Liver inactivation → Undergoes “first-pass effect”
Parenteral (injection) administration
fast (no need to wait for the absorption process via the intestines):
o Bypasses the liver (accurate); no first-pass effect
o Sterility is crucial when administering injection to prevent infection
Inhalation administration
o Rapid absorption through the lungs due to the large surface area of alveoli and high capillary exchange.
o Ideally, inhaled drugs only affect the lungs.
o However, they can have many sites of action if: They are absorbed by the vascular system in the lungs or some of the drug was swallowed and absorbed by the GI tract (depends on bioavailability)
o Systemic effects are rapid because if the drug is absorbed by the vascular system in the lungs, it bypasses the liver, so no first-pass (example: nicotine or sniffing cocaine)
Sublingual administration
rapid, no first pass:
o absorbed rapidly due to high blood flow of the mouth + bypasses the liver (no first-pass effect)
Absorption
• The drug administered needs to enter the bloodstream unless it's already there (ex., injected intravenously)
• Drugs administered orally, intramuscularly, or subcutaneously must cross plasma membranes to be absorbed and enter the systemic circulation. Tight junctions exist between cells, so a drug must cross the cell (not slip in between)
Mechanisms of absorption
Passive diffusion / facilitated diffusion (used by most drugs, no energy required, lipid-soluble drugs diffuse easily).
Active transport (e.g., glucose, amino acids; requires energy).
Sites of Absorption
Stomach: acidic
Small intestine: basic
Liver
Routes of administration & absorption patterns
IV (intravenous): 100% bioavailability, rapid peak, but irreversible.
IM (intramuscular): slower absorption, lower peak than IV.
Sublingual: bypasses liver, fairly rapid absorption.
Oral (swallowed): slowest, lowest peak; subject to first-pass effect.
Rectal: variable; some blood bypasses liver, some do not.
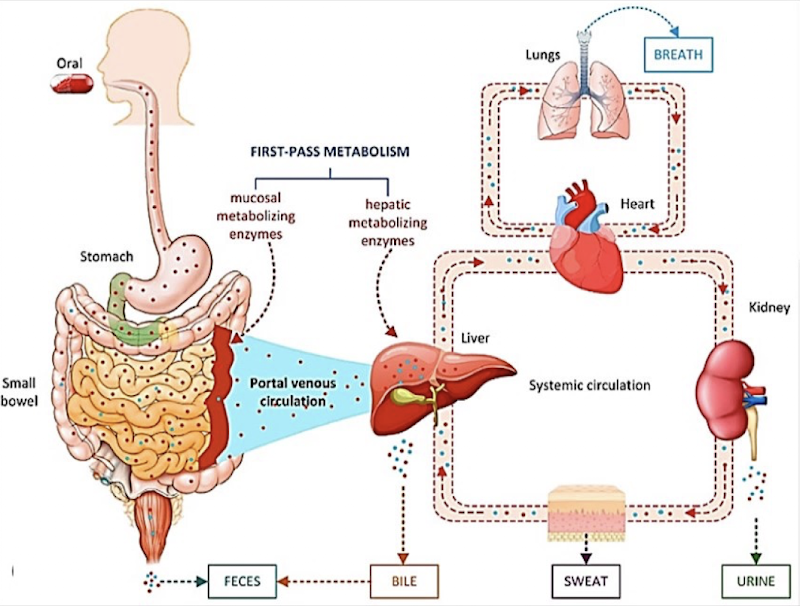
First-Pass Metabolism
Drugs taken enterally (e.g., oral) may be extensively metabolized in the liver before reaching circulation.
If most of the drug is destroyed, → need very high doses → risk of toxicity (from drug or metabolites).
Some drugs must be given IV because the oral route won’t achieve effective levels.
Distribution
o Drugs exist in two forms in the blood: protein-bound & free drug
o Only the free drug can leave the circulation and enter tissues, cross cell membranes, etc.
o Inside the tissues, the free drug can bind to intracellular proteins (reversible) + can get stored in fat → acts as a reservoir
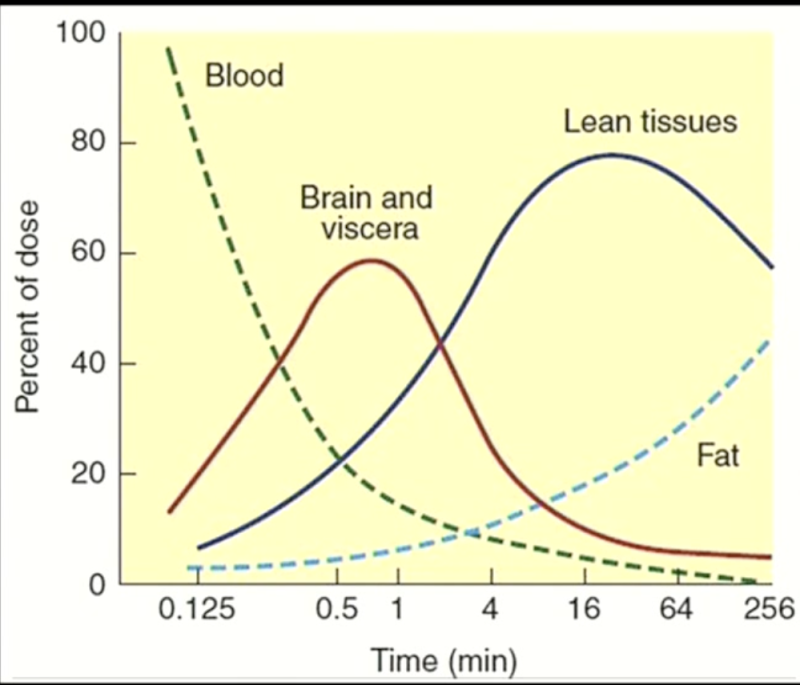
o The concentration of the drug FIRST goes up in the brain—a vessel-rich group. The brain, heart, liver and kidney have a very high fraction of the cardiac output.
o As the concentration of the drug is declining in the circulation, the concentration of the drug is quickly going up in tissues that are getting the highest blood flow (vessel-rich groups).
o Fat has a slower blood flow than vessel-rich groups and muscle groups.
Factors affecting the drug permeability
Free drugs that are lipid-soluble can go right through the capillary wall. Non-fat-soluble drugs use active transport to enter cells.
Peripheral capillaries: leaky, easy for drugs to leave the blood.
Brain capillaries (blood-brain barrier): Endothelial cells are tightly joined → no pores. Thus, only lipid-soluble drugs or those with special transporters can enter.
Is the placenta a good barrier for drugs?
• Is NOT an effective barrier to drugs
• Lipid-soluble drugs very rapidly diffuse through the placenta, and even water-soluble drugs can get through slowly.
• The implication, then, is that some drugs can concentrate in the fetus. Caution needs to be taken during pregnancy.
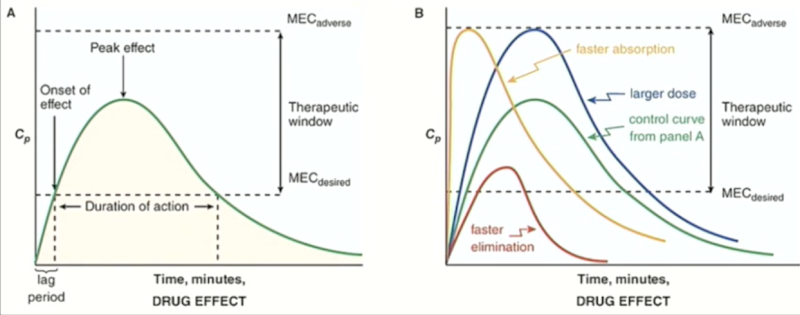
Explain the phases of Drug effect
•Lag period: blood concentration must reach a minimum effective concentration (MEC) before the drug works.
• Duration of action: Time during which blood levels are above MEC.
•Eventually, concentration falls below MEC → drug stops being effective.
•Therapeutic window: Range between MEC for desired response and MEC for adverse response
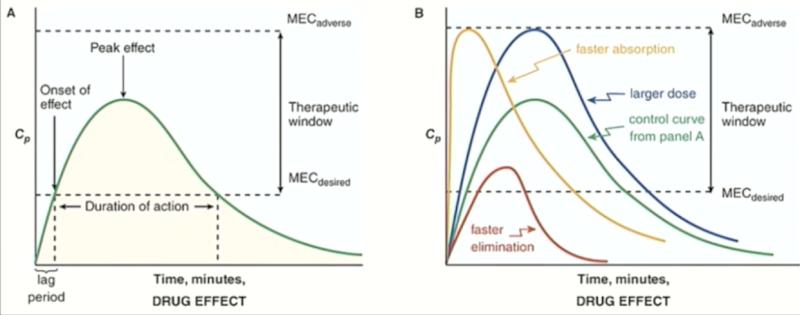
Variables affecting drug concentration
o The green curve is the control, the ideal drug.
o If we increase the dose, the curve (blue) goes up to the toxic effects, which is dangerous.
o For a drug that is absorbed quickly (yellow), the levels rise fast, but so does the risk of experiencing toxic effects
o The drug that is eliminated quickly due to fast metabolism of the drug (red) is also not ideal, as it does not stay above the threshold for long, so will not have effects for long.
Protein Binding in Circulation
• Drugs bind reversibly to plasma proteins such as albumin (major one) and α₁-acid glycoprotein.
• While bound, the drug is inactive (can’t leave the circulation, can’t be metabolized, can’t act).
• Plasma proteins themselves are too large to leave the vasculature, which is why bound drug is trapped.
How can a disease alter plasma binding in circulation?
o Albumin ↓ in liver disease → more free drug
o α₁-acid glycoprotein ↑ in inflammation → less free drug.
o Drug–drug displacement from protein binding can happen, but is rare and usually clinically minor unless the drug has a narrow therapeutic index (e.g., warfarin).
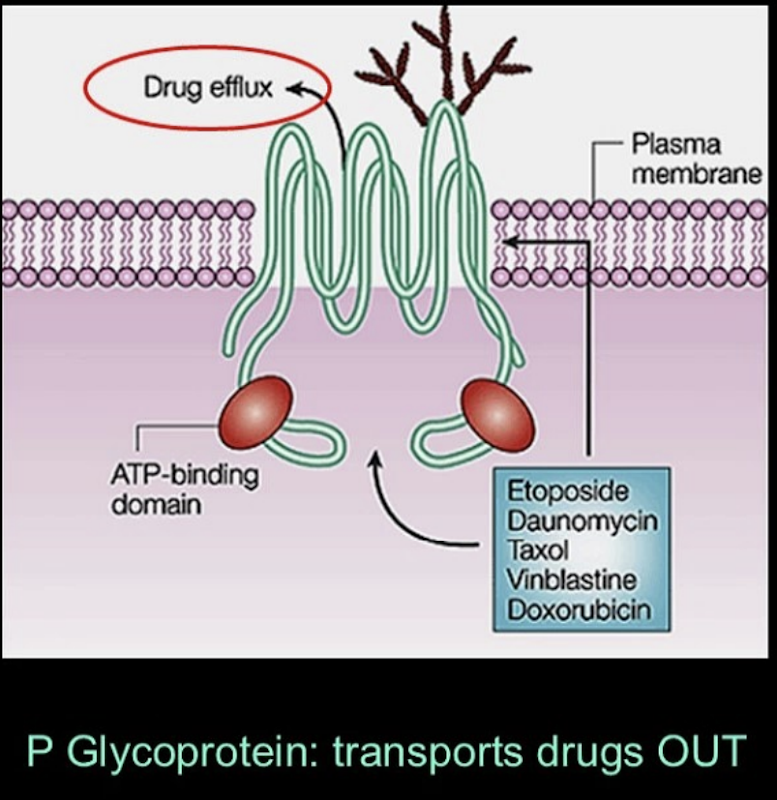
Cell-Level Transport
•Cells aren’t passive; they can control drug entry and exit.
•P-glycoprotein is an important “drug efflux pump”:
o Found in intestines and often overexpressed in cancer cells.
o Kicks drugs back out of the cell, reducing their effectiveness.
•Different tissues (intestine, liver, kidney, brain) have different sets of transporters, which affect how drugs move.
•Diseases can change transporter function, altering drug distribution.
Apparent volume of distribution (AVD or Vd)
Theoretical volume of a drug that would make the drug concentration equal throughout the body and plasma:
The drug ends up in the blood (central volume) soon after it is administered. Over time, it is also distributed in the periphery (peripheral volume).
Distribution is rarely uniform. Depending on drug properties, more may stay in the blood or move into peripheral tissues
⇒ the volume of distribution is apparent.
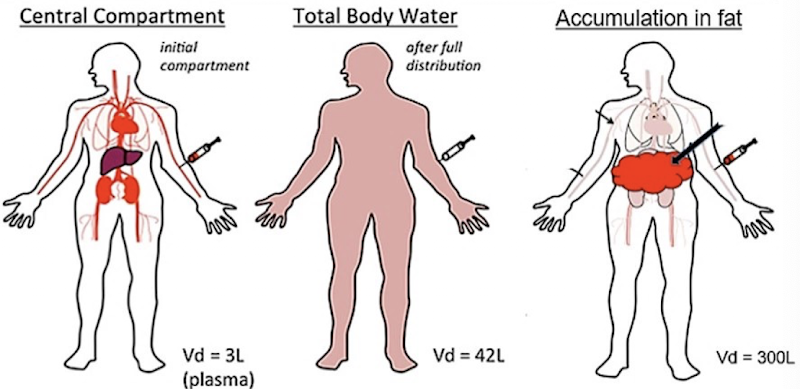
Major variables affecting AVD
Binding to plasma proteins or tissue proteins
Absorption into adipose tissue
Drug lipophilicity/hydrophilicity and MW
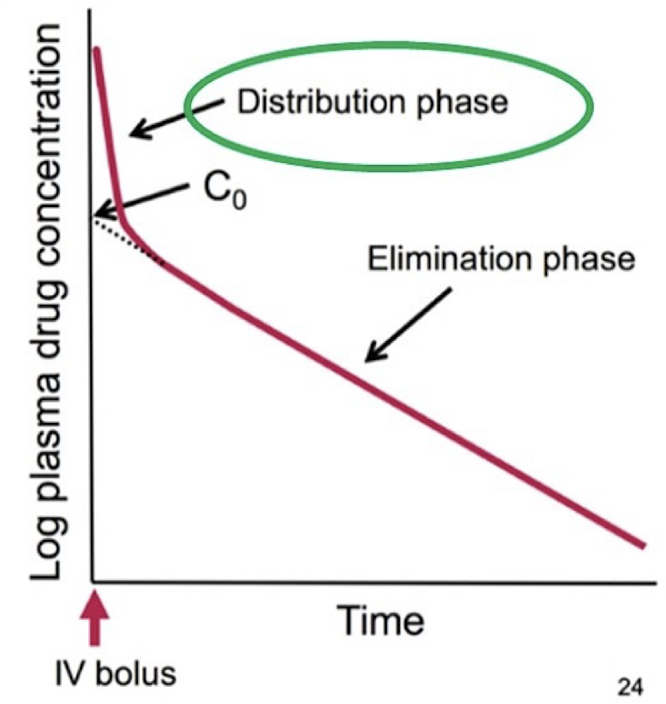
C0 (Cp or Cplasma)
Initial plasma concentration of the drug. Obtained by extrapolating the elimination phase back to t = 0 (when the drug is first administered). This is easiest to measure after IV administration, since the drug enters the plasma immediately (no first-pass).
AVD equation
Amount of drug in body = dose given
Concentration of drug in circulation = C0 if distributed immediately

In what cases will we have a high and low AVD?
Plasma protein binding → [Drug]plasma is high → LOW AVD
Tissue binding → [Drug]plasma is low → HIGH AVD
![<ul><li><p><span>Plasma protein binding → [Drug]</span><span><sub><span>plasma</span></sub><span> is high → LOW AVD</span></span></p></li><li><p><span>Tissue binding → [Drug]</span><span><sub><span>plasma</span></sub><span> is low → HIGH AVD</span></span></p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/13891b95-1e39-48fb-9985-e1da2ef7e792.png)
Metabolism
Conversion of a drug to less active or inactive metabolite(s):
The change generally involves the conversion of a nonpolar, lipid-soluble compound to a more polar form that is more water-soluble and can be more readily excreted in the urine.
Sometimes converts an inactive prodrug to its active form.
Sites of metabolism
Metabolism occurs mainly in the liver, but there are also various drug metabolizing enzymes in the skin, intestinal walls, kidneys, lungs, and brain:
Skin and lungs: to protect us when we inhale or come in contact with toxic substances.
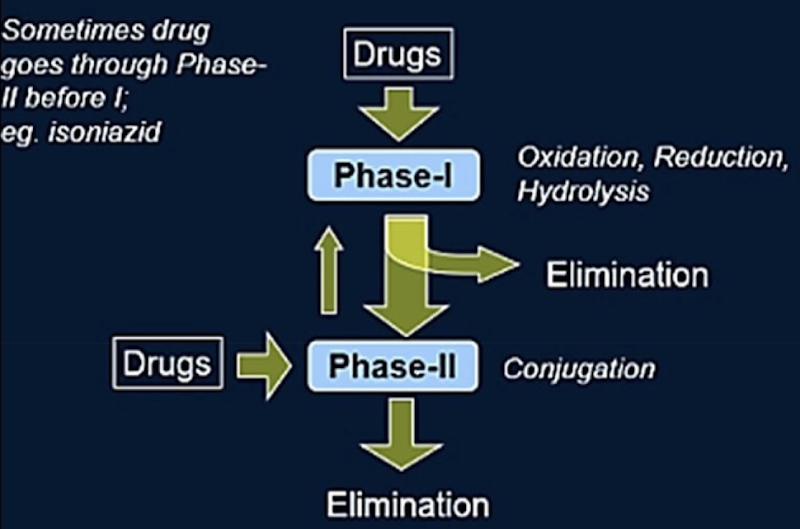
Phases of drug metabolism
Phase I: Oxidation/Reduction/Hydrolysis
Mainly occurs in the liver (+ in the intestine, lungs, kidneys and skin)
Carried out mainly by Cytochrome P450 enzymes
Main reactions are oxidation, reduction and hydrolysis
Usually inactivates the drug, but sometimes produces an active metabolite (ex.: prodrugs)
Phase II: Conjugation
Adds something onto the metabolite to make it water-soluble so the kidney can excrete it.
Carried out by transferases
Phase II products are generally inactive and easily eliminated, though occasionally an active metabolite can be formed.
Not all drugs necessarily go through Phase I followed by Phase II.
CYP450 in phase I elimination
Cytochrome P450 is a group of enzymes responsible for most reactions in this phase of drug metabolism.
Most are located in the liver (made in smooth endoplasmic reticulum of hepatocytes), but some are also found in the GI tract, the lungs, the skin and the kidneys.
First-pass metabolism mostly takes place in the liver, but some breakdown occurs in the walls of the intestine, before the drug even reaches the liver.
In cases of liver injury, the first-pass mechanism is altered, and the dose must be adjusted.
P450 enzymes are essential for survival. Their number has grown throughout evolution.
Nomenclature of the CYP family
Cytochrome P450: CYP
Family: number 1-17
Sub-family: letter
Enzyme or gene: number

Bioavailability
Percentage of the original dose of a drug that actually enters the circulation (following the first pass).
How can genetic factors alter drug metabolism?
Poor metabolizers: Require a lower drug dose to avoid toxicity. Prevent administering subsequent doses too quickly, or the drug level might rise above toxic levels quickly.
Effective metabolizers: Average responders to a drug. Ultrarapid metabolizers: Require larger doses to get an effective response. Drug concentration decreases quickly → frequent doses.
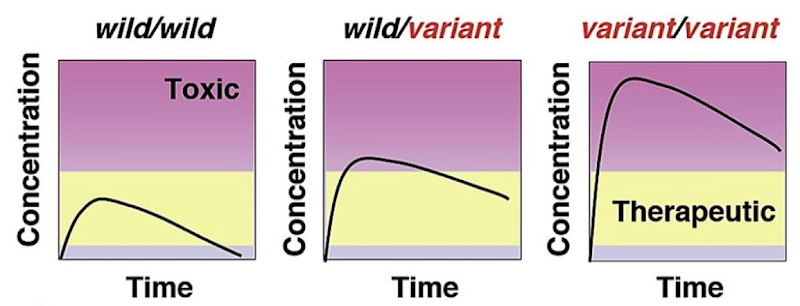
How can environmental factors influence drug metabolism?
Drug therapy – Drugs can alter the drug metabolism of other drugs (induce/inhibit enzymatic activity). Drug interactions.
Liver damage – Affects enzymatic activity. Often caused by excessive alcohol consumption.
Nutrition – dietary factors, starvation, available foods
Fever, stress and infection
How can age impact metabolism
Infants cannot metabolize drugs properly because they don’t have the full complement of mature drug-metabolizing enzymes yet
There is a tendency for drug metabolism to decrease with age, but there are exceptions
As a precaution, elderly people are given lower doses of a drug and their effects are monitored, and the dosage is increased as necessary.
Transferases
Enzymes that catalyze Phase II reactions. They transfer a conjugate onto the metabolite from Phase I to render it water soluble in the kidney for excretion.
Ex.:
GSTs transfer glutathione
UGTs transfer glucuronic acid
Liver’s role in metabolism
Main organ for metabolism (of all exogenous compounds)
Second largest organ after skin, greatest regenerative ability, vital for life
Large functional reserve → can increase its function when the body is under stress
Main site of first-pass metabolism:
Oral drugs are absorbed in the intestine → all portal blood flows through the liver first
Many drugs are partially inactivated during this first pass before reaching systemic circulation
Drug-metabolizing enzymes in hepatocytes:
Most Phase I (CYP450s) enzymes are located in the Smooth Endoplasmic Reticulum of hepatocytes.
Hepatocytes are in close contact with circulating blood, enabling efficient metabolism of substrates.
Liver disorders
Disorders are common and symptoms are diverse. They can affect drug metabolism.
A damaged liver is less capable of metabolizing drugs, which leads to higher plasma drug levels and an increased risk of toxicity.
Injury also reduces the liver’s ability to handle other drugs and its normal biological functions
In patients with liver disease, drugs are metabolized more slowly → dosage must be reduced.
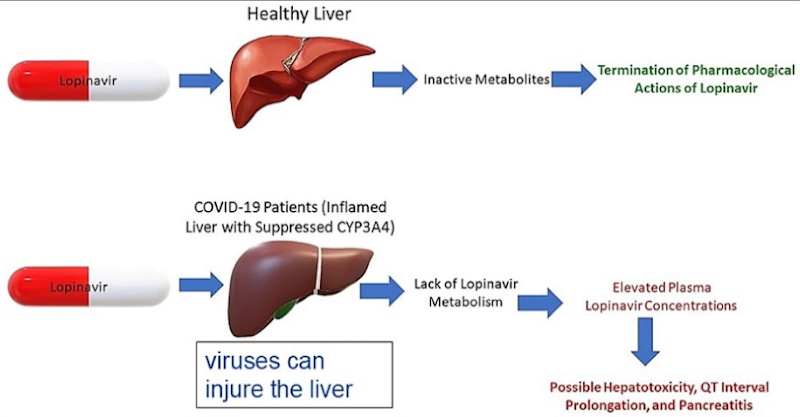
Causes of liver disorders
Chemical injury – alcohol, drugs, environmental toxins
Viral injury – infections like COVID-19 can alter enzyme activity
Ex.: Cirrhosis – potential fatal liver injury that can be caused by alcohol abuse and hepatitis
Kidney function?
Primary major organ of excretion
Eliminates both the parent drug (sometimes) and metabolites (often after Phase II conjugation)
Ex.: Aspirin
⅔ metabolized in liver → excreted in inactive form (renal elimination + hepatobiliary elimination)
⅓ renally excreted as an unchanged active substance in urine
Mechanisms of renal handling
Filtration: occurs at Bowman’s capsule:
Only free drug (unbound to albumin) can pass through
Albumin-bound drug is too large, so it remains in circulation until it converted to free drug form.
Dynamic equilibrium between bound and free drug → as free drug is filtered, more bound drug dissociates.
Secretion: drug can be transported from the blood into the proximal tubule (close connection between vasculature:
Reabsorption: substances taken back into the bloodstream:
Essential step because many filtered substances are essential to life.
Lipophilic filtered drugs can be reabsorbed back into the blood from the tubules. This is why water solubility is important for effective excretion (Phase II).
How does the liver work together with the kidney?
Liver metabolism makes drugs more water-soluble → the kidney can then efficiently eliminate them.
If the drug remains lipid-soluble, it may be reabsorbed in the proximal tubule and return to the liver.
Other forms of drug excretion (besides renal)
Biliary/fecal excretion: metabolites secreted into bile are eliminated by the intestine. Most is excreted in feces.
Lungs: volatile drugs and metabolites can be exhaled. Explains the smell of food on the breath.
Ex.: Ethanol – this is the basis for the breathalyzer test
Saliva: some drugs/metabolites are secreted into saliva. Explains the lingering smell of food.
Ex.: Smelling like garlic after a meal even if you brush your teeth.
Ex.: Saliva test for cannabis
Also, some excretion is possible through sweat.
Drug clearance
Drug clearance quantifies the elimination of a drug – the volume of body fluid cleared per unit of time. Normally a constant rate (L/hour, mL/min)
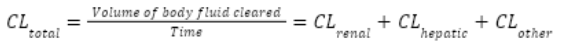
Renal clearance
filtration (GFR) + secretion - reabsorption
Hepatic clearance
the volume of blood the liver clears of drug per unit time, determined by liver blood flow and metabolic capacity (enzyme activity, first-pass effect).
How many half lives does it take to get rid of a drug?
4

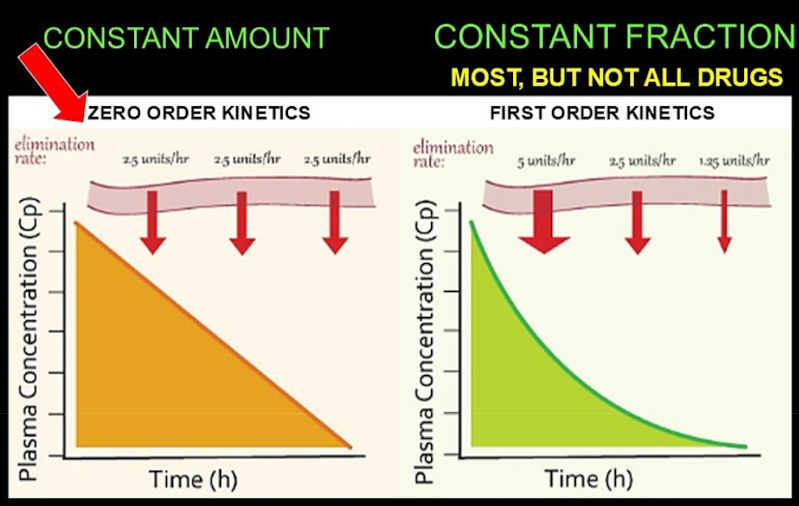
First vs zero order kinetics
First-order kinetics (exponential): Constant fraction per unit time:
Enough enzymes to metabolize larger amounts. No saturation.
Zero-order kinetics (linear): Constant amount of drug eliminated:
Saturation levels are reached for an enzyme → a constant amount of a drug can be processed.

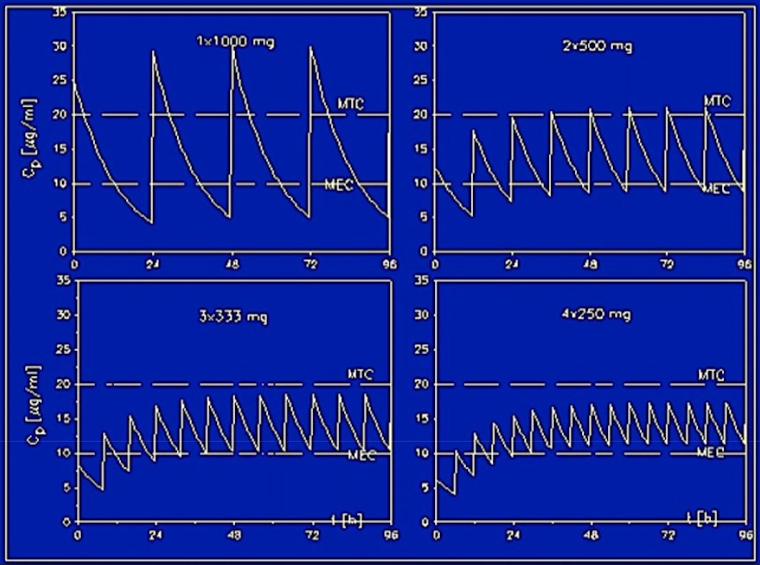
Dosing schedule
Since the duration of action is ~4 half-lives, a drug should be readministered at multiples of the half-life:
The drug concentration reaches a steady state after ~4 half-lives.
1st dose: 100%, drops to 50% after T1/2
2nd dose: 50% → 150%, drops to 75% after T1/2
3rd dose: 75% → 175%, drops to 87.5% after T1/2
4th dose: 87.5% → 187.5%, drops to 93.75% after T1/2
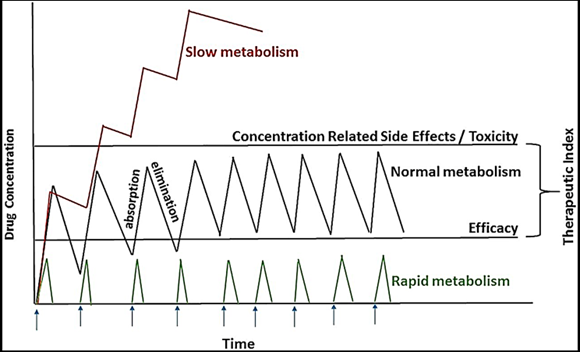
How do long and short half-lives impact the dosing schedule?
Short half-lives = more frequent administration
Long half-lives = less frequent administration

How do genetic differences affect dosing schedules?
Ultrarapid metabolizers may need more frequent and/or higher doses
Poor metabolizers may need less frequent and/or lower doses.
Poorly calculated dosing schedules could cause toxicity in poor metabolizers.
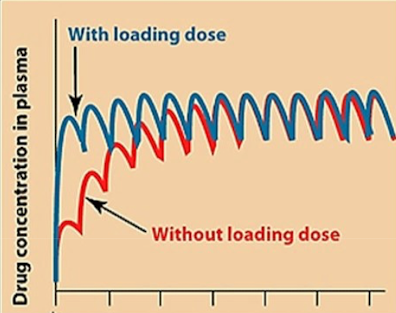
In emergencies, how do we quickly get to steady state?
For emergencies, a higher “loading” dose is administered first, followed by “maintenance” doses, to reach the steady state faster.
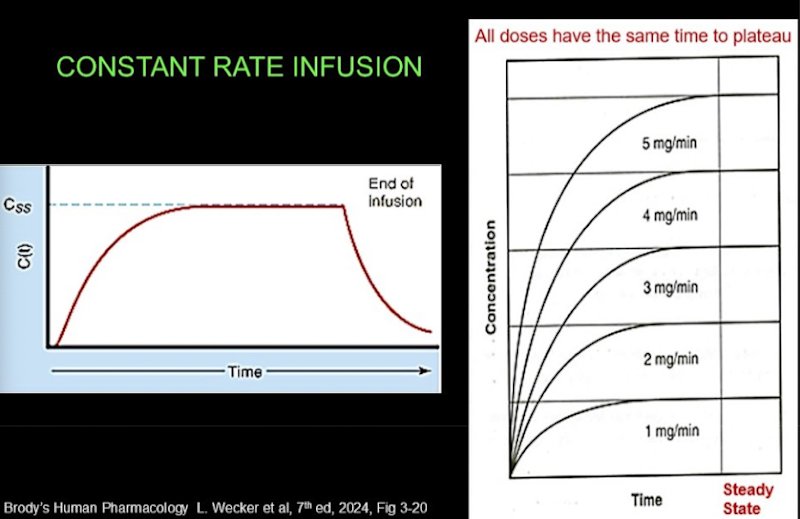
Constant rate infusion
When a drug is infused, plasma drug concentration rises gradually until it reaches a steady state:
Time to steady state is the same regardless of infusion rate: it depends only on the half-life.
What’s the site of drug excretion?
The kidney is a key site for the elimination of endogenous and exogenous substances and includes glomerular filtration, tubular secretion, and tubular reabsorption
Concentrations in the kidneys must be monitored to avoid damage during elimination.

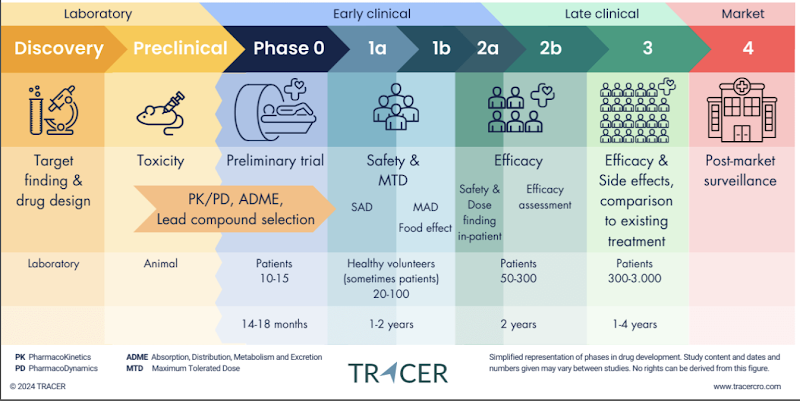
Drug Development Process
Target validation: find a molecule (e.g., antibody, small drug) that can block or stimulate a receptor/pathway.
Preclinical (animals) & Phase 0: early pharmacokinetics (how drug moves through body).
Phase 1: give drug to healthy volunteers, mainly to check safety and toxicity. Some pharmacokinetics is measured too (how long it lasts, how it’s distributed).
Why Drugs Fail
Usually a pharmacokinetic problem:
Even if a drug works well on the target, it can fail because of how the body handles it.
Pharmacokinetics failures (ADME: absorption, distribution, metabolism, excretion).
Examples: short half-life, precipitation, drug accumulates in the wrong organ
Adverse Drug Related Hospitalizations (ADRs) vs Individual Variability in Drug Response (IVDR)
ADR: ADR refers to harmful reactions to a drug.
IVDR: IVDR refers to differences in how individuals respond to the same drug
Factors Influencing IVDR
Individual variability is influenced by physiological and pathological processes that are associated with specific pharmacokinetics and pharmacodynamic properties (usually referred to as parameters) of the drug.
Physiological processes
Water content, fat content, mineral composition in females vs males can affect drug response.
Body size, maturation of organ function in infants
Pathologic processes (ex: heart failure, renal failure) may be used for dosage adjustment in individual patients.
How does age affect drugs
Drug metabolism differs based on age.
Infants, young adults, and elderly individuals metabolize drugs differently due to factors such as body size and organ function.
Babies and children are NOT just miniature adults!
An infant will metabolize and store a drug differently from an adult because of the size of the infant
Clinical trials typically focus on young adults, and the effects on other age groups are sometimes neglected.
Age differences are often not thought about in clinical trials: they are usually done on young adults.
Infants present a unique challenge because their bodies differ more significantly from adults than elderly individuals.