Hematopoietic System
1/55
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
56 Terms
Equation Note
% in the equations means in actual percent, like 0.01 instead of 1
Terms for Formation of Blood
Hematopoiesis - formation of blood
Erythropoiesis - formation of red blood cells
Leukopoiesis - formation of white blood cells
Functions of the Blood
Transport: transportation of nutrients, O2, hormones, wastes (nitrogenous wastes,CO2), heat
Regulation: of pH, temperature, water content
Protection: from pathogens (phagocytosis, complement, interleukins, antibodies), bleeding (clotting system)
Blood Usual Physical Characteristics
pH: 7.35 to 7.45
Temperature: 38 Celsius
Centrifuged Blood Layers
Yellow Layer: Plasma
White Layer: Buffy Coat (Platelets and WBCs)
Red Layer: Red Blood Cells
Blood Composition
8% of total body mass
Splits into:
Plasma (55%):
91.5% water
1.5% solutes (gases, electrolytes, nutrients, wastes)
7% proteins (albumins, globulins, fibrinogen, others)
Formed Elements (45%):
Red Blood Cells (Most)
Platelets
White Blood Cells (Least)
Medical Products of Plasma
Fresh Frozen Plasma (FFP):
collected from whole blood donations
centrifuged, supernatant collected
frozen within 8 hours of collection
uses: prevention/stopping bleeding (through plasma clotting factors), and plasma donation
Cryoprecipitate:
FFP that is thawed, further centrifuged
insoluble precipitate is resuspended in liquid plasma
mostly concentrated fibrinogen and clotting factors
Hematopoiesis
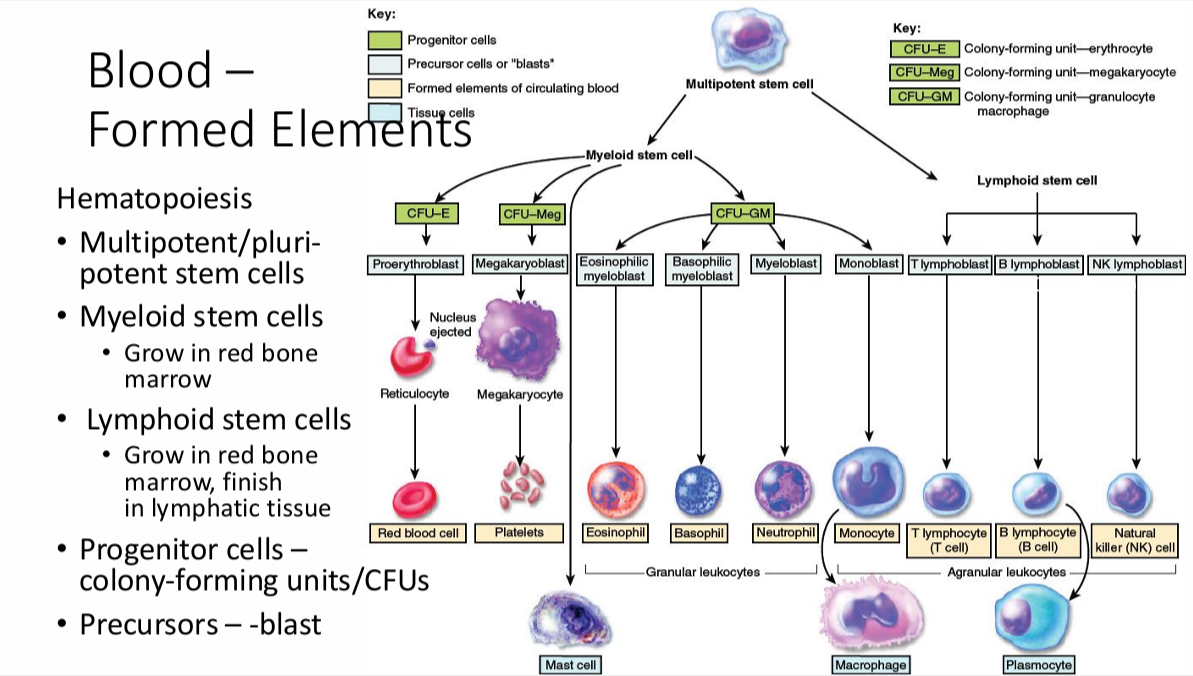
note: Reticulocytes are already in circulation
Growth Factors
Erythropoietin (EPO): increases RBC formation, note that testosterone increases EPO
Thrombopoietin (TPO): increases Megakaryocyte → platelet formation
Colony Stimulating Factor (CSF):
GM-CSF: for granulocytes and macrophages
Cytokines:
interleukins, chemokines, interferons, and tumor necrosis factors
also have other uses in the body
Stem Cell Therapy Indications
Hematopoietic Stem Cell Transplantation:
for patients with certain types of leukemia
after radiation kills bone marrow, stem cells are implanted to replace
Corneal Resurfacing:
corneal injuries are healed by transplant of corneal stem cells (usually from a recently dead donor)
Skin Regeneration with Epidermal Stem Cells:
for scars, burns, skin injuries
Life Cycle of a Red Blood Cells
Lifespan: around 120 days (3-4 months)
after which, it is too damaged and is phagocytosed
degrades into heme and globin and iron
Heme Pathway:
heme → biliverdin → bilirubin, gets stored in the liver
bilirubin → stercobilin (in feces through bile, makes poop brown)
bilirubin → urobilin (in urine through circulation, makes urine yellow)
Note: acholic stools are characteristic of bile blockage due to this
Iron Pathway:
when heme is broken down iron is released
hepcidin prevents iron reabsorption and prevents oxidation of Fe2+ to Fe3+ if iron is too high
Transferrin takes iron (in ferric state) to liver where it’s stored in ferritin
Globin Pathway:
broken down into amino acids to be reused for protein synthesis
Hemoglobin and Hemoglobin Characteristics
a protein and pigment
Normal Levels:
Male: 13-18 g/dL
Female: 12-16 g/dL
Each hemoglobin has four hemes each with one Fe2+ each, which can bind one O2 each
Each hemoglobin has two alpha + two beta/delta/gamma globin chains
Hemoglobin Types
A1: most common (95-98%), has 2 beta chains
A2: (2-3%), has 2 delta chains
F: fetal, (<1%), 2 gamma chains, can extract oxygen stronger to compete with mother for oxygen in the blood
Hemoglobin Disorders
Methemoglobinemia: inherited or acquired
Fe2+ → Fe3+ in heme, changing oxygen affinity, leading to decreased oxygen release
bluer skin
Thalassemia: genetic mutations that leads to underproduction/absence of globin chains
Alpha Thalassemia: no alpha chains, lethal, immediate death because alpha chains are in all
Beta Thalassemia: no beta chains, less lethal, lives but with conditions, causes microcytic, hypochromic RBCs
can either be no/less/altered beta-globin chains
causes anemia, hemolysis, extramedullary hematopoiesis (outside bone marrow), iron overload, etc.
Sickle Cell Disease:
HbA1 → HbS hemoglobin
HbS polymerizes, forming abnormal cell structure (crescent shape instead of biconcave)
tends to be immune to some malarias, weirdly
prone to hemolysis, vasculopathy (abnormal blood vessels), priapism (sustained painful erection), chest pain
Erythrocyte Parameters 1
Hemoglobin Levels
Red Blood Cell Count
RBC characteristics (shape, color, size, parasite presence with peripheral blood smear)
Terms: micro/macrocytic for size, hyper/hypochromic for color
Hematocrit
% of RBC volume in centrifuged blood
high = polycythemia, dehydration
low = anemia
Reticulocyte Count (used as a surrogate for hematopoiesis rate)
normal level 0.5-1.5%
Erythrocyte Parameters 2
Mean Corpuscular Volume (MCV)
formula: (Hematocrit% x 10)/RBC Count (in millions/mm3)
Normal: 80-100 fL (femtoliters)
Mean Cell Hemoglobin (MCH)
formula: Hgb count/RBC count
normal: 27-31 picograms/cell
Mean Corpuscular Hemoglobin Concentration (MCHC)
hemoglobin per volume of RBCs
normochromic = 32-36 g/dL
vs hyper/hypochromic
Erythrocyte Sedimentation Rate
marker of inflammation
sedimentation rate of RBCs in vertical tube
increased sedimentation with increased aggregation due to clotting factors
too slow = not enough clotting, too fast = too much clotting
Anemias
Definition: a deficiency of either RBCs or hemoglobin
Hemoglobin Disorders (can cause anemia)
Iron Deficiency Anemia
microcytic, hypochromic
causes: reduced iron intake (vegetarian, insecure food source), bleeding, increased iron requirements (childhood, pregnancy)
Diagnosis: low Transferrin Saturation (TSAT) or low Ferritin
Pernicious Anemia
macrocytic/megaloblastic
caused by vitamin B12 deficiency
causes: usually intrinsic factor deficiency → lower B12 absorption
Hemolytic Anemia
premature RBC rupturing leads to less blood cells
due to many conditions (ex: sickle cell, hemolytic disease of newborn, etc.)
Aplastic Anemia
bone marrow does not meet RBC production demand
Blood in Feces
Melena:
some blood in feces
black feces, muddy consistency
Hematochezia:
significant blood in feces
red blood visibly in feces, or blood coming out of anus
A Yoshiko for your troubles

Differential White Blood Cell Count
the individual count of each different type of leukocyte
in the past, used a handheld device but these days it is done mostly electronically
Leukocyte Types
Granular Leukocytes:
appears to have granules under microscope
Neutrophil
Eosinophil
Basophil
Agranular Leukocytes:
still has granules but are not visible under light microscopy
Lymphocytes
Monocytes
Neutrophils
the most common white blood cell
nucleus has multiple lobes
also called polymorphonuclear leukocytes (PMNs) or polys
Eliminate pathogens via phagocytosis
High Count: indicator of infection, burns, stress, inflammation
Low Count: indicator of radiation exposure, immune conditions (ex: lupus), or vitamin deficiency (ex: B12)
Neutrophil Migration
Steps:
Tethering: attaching to the wall of the blood vessel
Rolling: rolls across the wall to the spot it needs to go
Transmigration: slips between the spaces between endothelial cells
Neutrophil recognizes markers like E-selectin, Integrins, ICAM, and VCAM (adhesion molecules) as attachment points on the vessel walls
This lets neutrophil enter tissue to attack pathogens there
Also involved:
histamine
prostanoids
cytokines
which produce vasodilation
Absolute Neutrophil Count
Formula: WBC Count x %(PMN + Bands)
(PMN means polys or mature Neutrophils, while Bands are immature)
particularly important in cancer chemotherapy
Neutropenia
defined as a low neutrophil count
severity is based on Absolute Neutrophil Count (ANC)
Mild: ANC => 1000, but <1500 cells/μL
Moderate: ANC => 500, but <1000 cells/μL
Severe: ANC < 500 cells/μL
Severe tends to have patient having sudden and inexplicable fevers
Benign Ethnic Neutropenia (BEN):
congenital condition; persistently low neutrophil counts African, Middle Eastern, and West Indian descent
ANC < 1000-1200 cells/μL
Other Granulocytes
Eosinophils:
attacks parasites and allergies
stains red with acidic dyes
hence also called acidophils (?)
High Count: indicates allergic reaction or parasitic infection
Basophils:
important in immune and allergic response
stains blue with basic dyes
High Count: indicates some allergic reactions, some cancers, hypothyroidism
Lymphocytes
attacks viruses, leukemias, cancers, malignant cells, infectious mononucleosis
High Count: viral infection, leukemia, infectious mononucleosis
Low Count: prolonged illness, HIV infection, immunosuppression, treatment with cortisol
Function by recognizing antigens and inducing apoptosis
Chimeric Antigen Receptor (CAR)-T Cells
T Cells are collected from patients
Collected cells are exposed to specific viruses with specific antigens
T Cell then develops recognition for specific antigens
These T Cells are deployed against specific cancers (some leukemias)
Monocytes
become macrophages when they leave circulation
kidney or horseshoe shaped nucleus
High Count: viral & fungal infections, some leukemias, some chronic diseases
Low Count: bone marrow suppression, treatment with cortisol
Note: tuberculosis can stay dormant within the macrophage
Characteristics and Functions of Leukocytes:
deal with pathogens via phagocytosis or other immune response
typically live for only a few hours or few days (except lymphocytes that live for years, ex: memory cells)
once they leave circulation, they can not return (except lymphocytes)
High WBC Count: leukocytosis
Low WBC Count: leukopenia
Emigration (Details)
guided by adhesion molecules
Chemotaxis: phagocytotic cells are “attracted” to the correct areas via substances secreted during inflammation and infection
(ex: cytokines)
Nico Sass for Energy

Platelets
Megakaryoblast → Megakaryocyte → Platelets
each Megakaryocyte forms around 2000-3000 platelets
non-nucleated cell fragments
Normal Value: 150,000 - 450,000 platelets/μL
Irregular, and disc shaped
Important for hemostasis
Hemostasis
the process of controlling blood flow out of an injured blood vessel
or
a sequence of responses that stops bleeding (prevents hemorrhage, internal or external)
Mechanisms:
Vascular Spasm: contraction of blood vessels limits flow to damaged area
Platelet Plug Formation
Blood Clotting/Coagulation or Coagulation Cascade
Fibrinolysis
Platelet Plug Fomation
also called primary hemostasis
Steps:
Platelet Activation and Adhesion:
governed by adhesion receptors (ex: GPIIb/IIIa on platelet surface, GP = glycoprotein)
adhesion receptors bind to various ligands which then adhere to the receptors (ex: collagen, vWF or von Willebrand Factor)
Rationale: these ligands (ex: collagen) are only exposed when the vessels are damaged
other activators: Thrombin
Platelet Release Reaction:
after binding, platelets activate and release granules
Granules contain various molecules
ex: 5-HT, ADP, TXA2 contribute to further platelet activation
5-HT and ADP are also vasoconstrictive
TXA2 comes from COX pathway
Platelet Aggregation:
Fibrinogen binds to the GPIIb/IIIa receptor
Fibrinogen acts as a “rope” to stabilize platelets in place
Coagulation then takes place if necessary
Specific Roles of Molecules in Platelet Release Activation
ADP:
binds to purinergic P2Y12 receptors which activate platelets
5-HT:
binds to 5-HT2a receptors which further promotes granule release
synergistic with other agonists
can coat platelets via protein serotonylation
Potential Link of Primary Hemostasis to Secondary Hemostasis
procoagulant platelets exposing phosphatidylserine
promotion of generation of Factor Xa and Thrombin (Factor II)
Hypothetical, won’t come up in exam
Blood Clotting
secondary hemostasis
also called Coagulation
reinforces the platelet plug by creating a blood clot or thrombus
functions via a cascade of clotting factors
cascade is heavily reliant on calcium
Thrombosis: the abnormal formation of blood clots that can lead to stroke or heart attack
Coagulation Cascade: Extrinsic Pathway
fast response (from seconds to minutes)
started by tissue trauma
relies on factors outside the blood vessels that enter due to trauma
Tissue Factor (Factor III) or thromboplastin outside the vessels bonds with Factor VII (and activates it to Factor VIIa) (requires calcium)
the Factor III/VIIa complex activates Factor X to Factor Xa
Factor V is activated by calcium to Factor Va
Factor Va combines with Factor Xa to form Prothrombinase
Coagulation Cascade: Intrinsic Pathway
slower response (takes several minutes)
started by endothelial cell damage (inner lining of blood vessel)
Factor XII is activated via various stimuli (ex: collagen in vessel walls or phospholipids from damaged platelets)
Factor XIIa activates Factor XI, which activates Factor IX
vWF stabilized Factor VIIIa, which then binds to Factor IXa
The Factor VIIIa/IXa complex activates Factor X into Factor Xa
Factor Xa combines with Factor Va to form Prothrombinase
requires Calcium
Note: thrombin can further activate Factor 8 and 11
Coagulation Cascade: Common Pathway
Prothrombinase activates Prothrombin (Factor II) into Thrombin (Factor IIa)
One prothrombinase can activate around a thousand thrombin (thrombin burst)
Thrombin activates platelets
It also activates fibrinogen (Factor I) into fibrin (Factor Ia)
Fibrin can
Fibrin attaches to Factor XIII which secures the platelet plug further
The fibrin form strands and act as a “rope” securing the plug in place
Vitamin K
can be sourced from leafy greens
Vitamin K is required for the synthesis of Factors II, VII, IX, X (mmemonic: 1972) (9-10-7-2)
fat-soluble, absorbed via intestine if lipid absorption is normal
Disorders that slow lipid absorption = Vitamin K deficiency = uncontrolled bleeding
Clot Retraction
happens once clot has formed
fibrin clot “tightens”
pulls the edges of the injury together, stemming blood loss and expediting healing
Hemophilia
condition characterized by less than 5-10% of normal Clotting Factor activity
Hemophilia A: Classic - Deficiency of Factor VIII
Hemophilia B: Christmas Disease - Deficiency of Factor IX
Hemophilia C: Rosenthal Syndrome - Deficiency of Factor XI (rarer?)
Fibrinolysis
dissolves small, inappropriate clots
also dissolves clots from healed wounds
Plasminogen is an inactive plasma enzyme that is incorporated into the clot when it is formed
Body tissues and blood contain components that activate it into Plasmin (or fibrinolysin)
ex: Thrombin (IIa) and Tissue Plasminogen Activator (t-PA)
Plasmin dissolves fibrin threads
D-dimers
breakdown product of fibrin
elevated in venous thromboembolism and COVID-19 (marker for inflammation)
can also be a marker for old age, pregnancy, contraceptive use, exercise
used for ruling out the previous conditions (rather than diagnosis)
Other Mechanisms for Clot Regulation & Termination
Primary Hemostasis (Platelet Plug) Inhibitors: Nitric Oxide (NO) (vasodilator) and Prostaglandin I2/Prostacyclin (PGI2)
Secondary Hemostasis (Coagulation Cascade) Regulators:
antithrombin
heparin
heparan
activated protein C
protein S
Aplastic Anemia
rare bone marrow failure
aplastic means no more tissue formation
absence of blood cell formation
Causes:
direct damage (medicine, chemicals, pathogens)
Genetic (inherited or random mutation)
Autoimmune disorder
S/Sx:
Pancytopenia (meaning thrombocytopenia + leukopenia + anemia, all cell types down)
Hypocellular Bone Marrow
Bleeding (less platelet), Infection Prone (less WBCs), Fatigue (less RBCs)
Tx:
transplant
immunosuppression
Growth Factor administration (ex: thrombopoietin receptor agonist)
Airi Toaster Appreciation

Blood Groups and Blood Types
Antigens that determine blood type are called agglutinogens and are generally glycoproteins and glycolipids
Antibodies related to blood types: agglutinins
Blood Group: based on the presence of absence of specific agglutinogens
ex: ABO Blood Group, Rh Blood Group
Blood Type: based on the specific antigen present within a blood group
ex: AB, O, A; Rh+, Rh-
Incidence does vary among population groups
Incompatible Blood Transfusion
Agglutination: clumping, occurs when agglutinins bind to antigens on incompatible RBCs, causing them to cross-link
afterwards, the complex attracts complement, causing leaky RBCs and hemolysis
liberated hemoglobin can also damage kidneys by blocking filters
ABO Blood Group
Blood can express, A antigens, B antigens, both, or neither
Type A: A only
Type B: B only
Type AB: both (universal recipient, since produces no antibodies)
Type O: neither (universal donor, since doesn’t have antigens, but can only receive O blood)
Note: the term “universal” is a misnomer since Rh Blood group is still needed to be considered, as well as other factors
Rh Blood Group
originally discovered in Rhesus monkeys
Rh factor is the name of the antigen displayed
Rh+: most common, Rh factor antigen is present
Rh-: less common, Rh factor antigen is absent
Normally, blood does not contain anti-Rh antibodies even if Rh-
An Rh- person develops antibodies after receiving Rh+ blood, which will then cause agglutination if Rh+ blood is given again
Proper Blood Screening
For ~80% of the population, ABO antibodies are present in saliva and other fluids which can be used in screening
blood is either cross-matched to potential donor blood, or blood is screened for antibody presence
for screening, drops are mixed with antisera (solutions containing antibodies) to observe reactions
after screening, cross-matching occurs
cross-matching: possible donor RBCs are mixed into patient serum to check reactions (or patient serum is checked against a test panel of RBCs)
Genetic Determination of Blood Type
alleles code for specific antigens
A and B are dominant, O is less dominant
So having A/B and O will result in A/B
having both A and B will result in AB
To get O, one must have O and O
Hemolytic Disease of the Newborn
blood of fetus and mother rarely come into contact, but it is possible through placenta
if Rh+ baby blood leaks into Rh- mother blood, mother will produce antibodies that attack baby
since blood leakage is most probably during delivery, first baby is usually unaffected
if mother has another Rh+ baby, the child becomes at risk
anti-Rh gamma globulin can help mitigate effects if used correctly
note: ABO agglutinins tend to be too large to cross placenta, so mother-infant ABO incompatibility rarely causes problems