BCH261 - Module 5 - Protein Structure
1/101
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
102 Terms
How is X-ray diffraction used to determine protein structure
models diffractions into an electron density map
Can measure distances between bonds
used by Linus Pauling
What were the rules discovered by X-ray diffraction?
1. Ca atom has tetrahedral
2. Carbonyl C has trigonal
3. Peptide bond is a double bond (between the carbonyl C and amino N)
4. But, this bond measured 1.32 A, which is shorter than a typical single bond but longer than a double bond
- this is due to partial double bond character due to resonance
What is special about peptide bond at the primary sequence level?
it is a resonance hybrid
- bond length is intermediate between single and double bond
- rotation about this bond is restricted and stiff (everything bends around it)
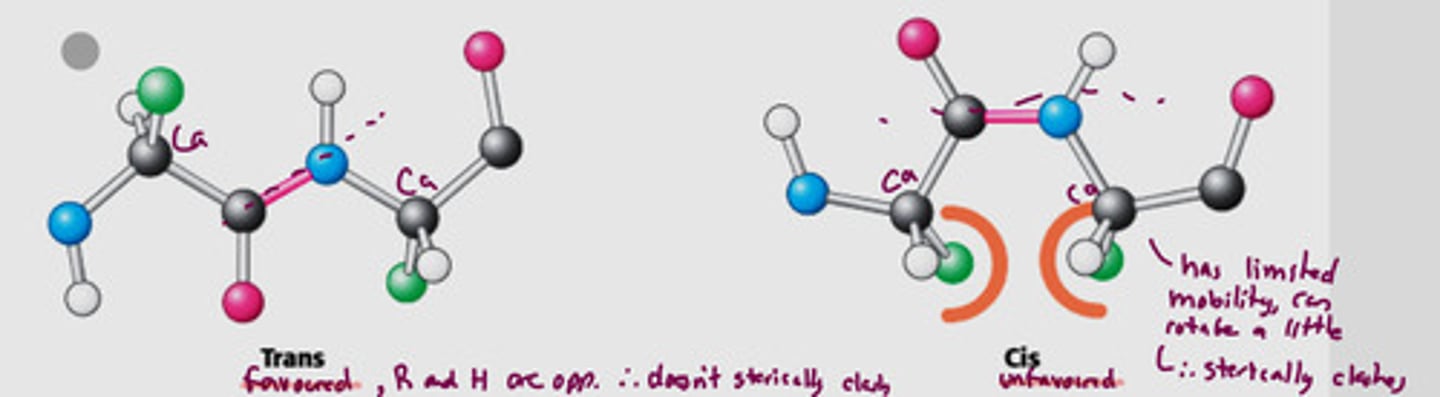
What two conformations are possible for a planar peptide bond? Which is more common?
Trans
- two alpha-C are on opposite sides of the peptide bond
- favoured, does not sterically clash
Cis
- two alpha-C are on the same side of the peptide bond
- unfavoured, sterically clashed, limited mobility
Trans is more common
What can rotation of bonds in peptide be described as?
two angles since bond is planar and trans
1. phi (between N and Ca)
2. psi (between Ca and C from carbonyl)
3. (not covered) omega (between N and C carbonyl) - 180 deg and rarely zero (since if it was zero it would not be trans)
Where is rotation allowed about
only about the N-Ca and Ca-C carbonyl bonds
Explain the freedom of rotation about bonds in a polypeptide
rotation about the phi and psi bonds are free (these are single bonds)
freedom of rotation about the two bonds of each amino acid allows proteins to fold in many different ways
What are the bond angles for phi and psi
phi = φ = -80 deg
psi = ψ = +85 deg
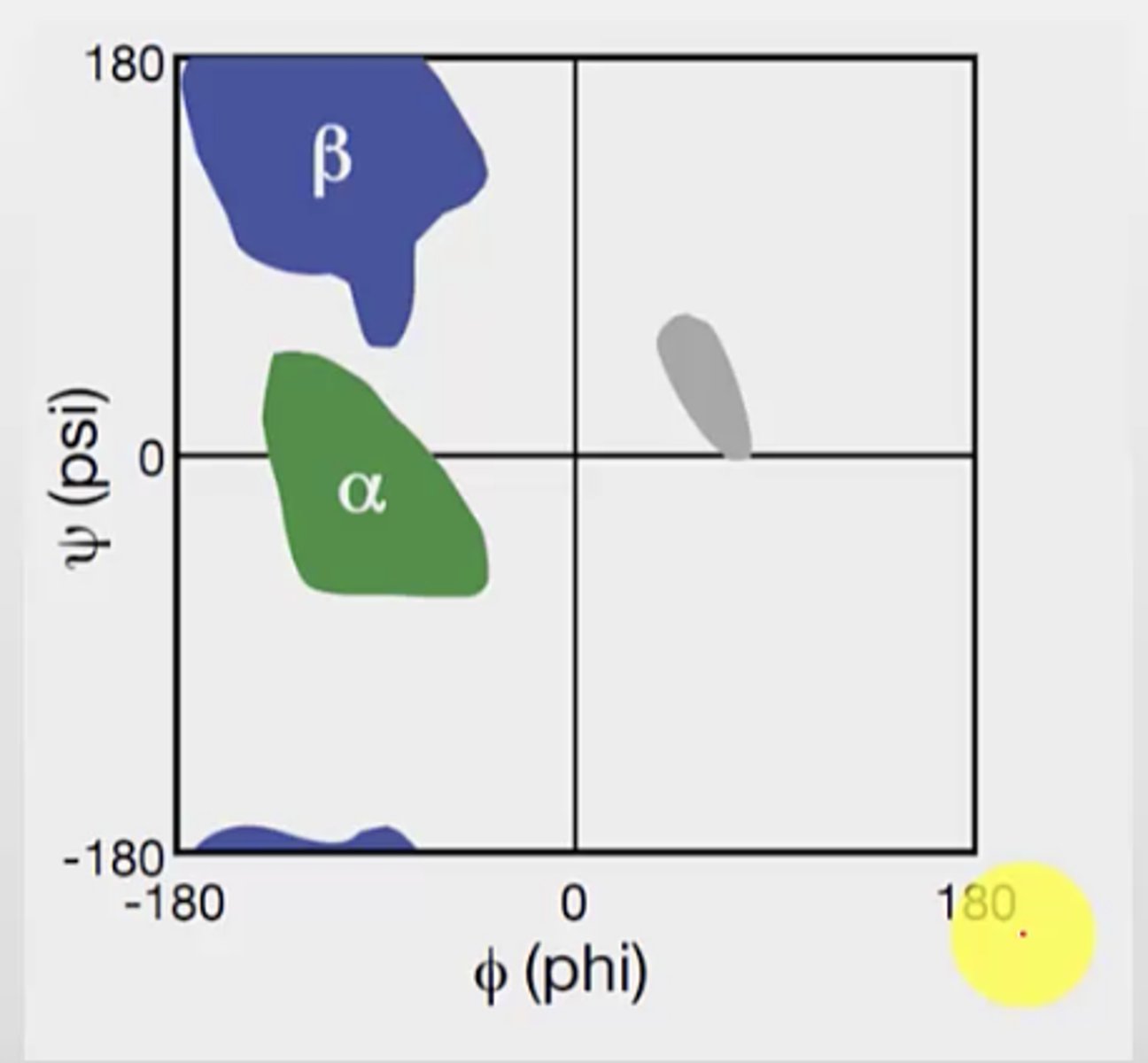
What does the Ramachandran plot show
distribution of phi and psi dihedral angles (angle between two planes/bonds) that are found in a protein
angle degree and direction (+ or -) is considered
x axis = phi
y axis = psi
Does the Ramachandran plot show every single bond angle possible? Why?
No, it does not show every single bond angle - this is because not every dihedral angle is possible in nature due to steric clashes
Only three regions of the plot has plotted points
The plot is based on bond rotation and hinderance discovered as the bond rotates
What are the most favourable quadrants on the plot? What is the structure of the peptide bonds at these locations?
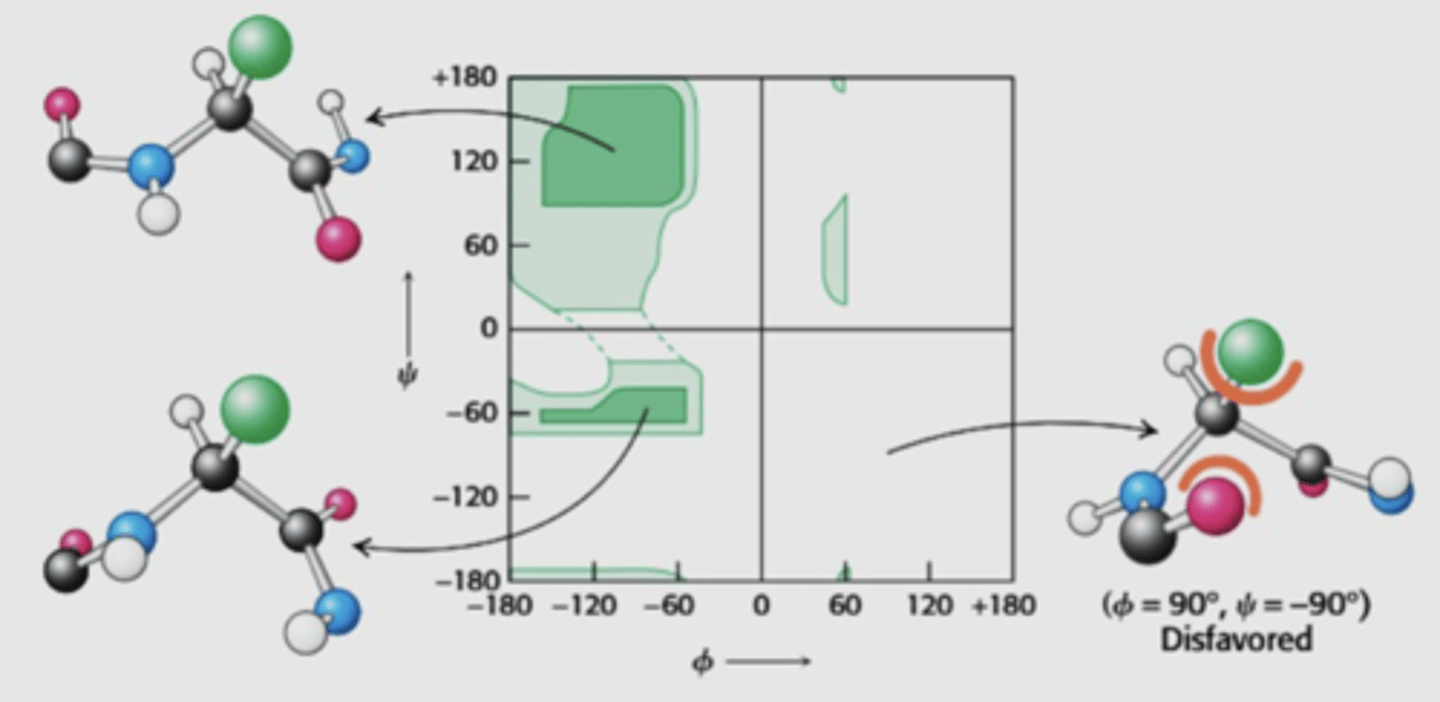
How does protein folding/structure depend on psi/phi angles
The rigidity of the peptide unit and the restricted set of allowed phi and psi angles limits the allowed structures of the protein
Only certain structures/folded forms of the protein is allowed due to limited angles allowed
What is the bond angles for alpha helix? What is special about alpha-helix?
phi = -57 deg
psi = -47 deg
Not set angles, they can vary
Special since it is the most common bond angles and the most commonly seen structure in proteins
Alpha helix resides between the 2nd and 3rd quadrants (most red part)
How are structures in secondary protein structures stable?
Why do unstructured regions happen?
Formed by non-covalent bonds
Stable due to hydrogen bonds and minimized steric hinderance
Unstructured regions are result of bond angles not repeated
What are the two forms of alpha helix?
1. Left-handed - rare (occupy region in quadrant 1)
2. Right-handed - common (occupy region in quadrant 3)
- more favourable (since less steric clash especially with L-amino acids)
- imagine a staircase you are going up from the bottom, if the stairs start on the right side - right-handed helix

How are H-bonds located between the turns of the helix
the carbonyl on one turn will form a hydrogen bond with the amino from the turn right above
look at slides for another perspective on this
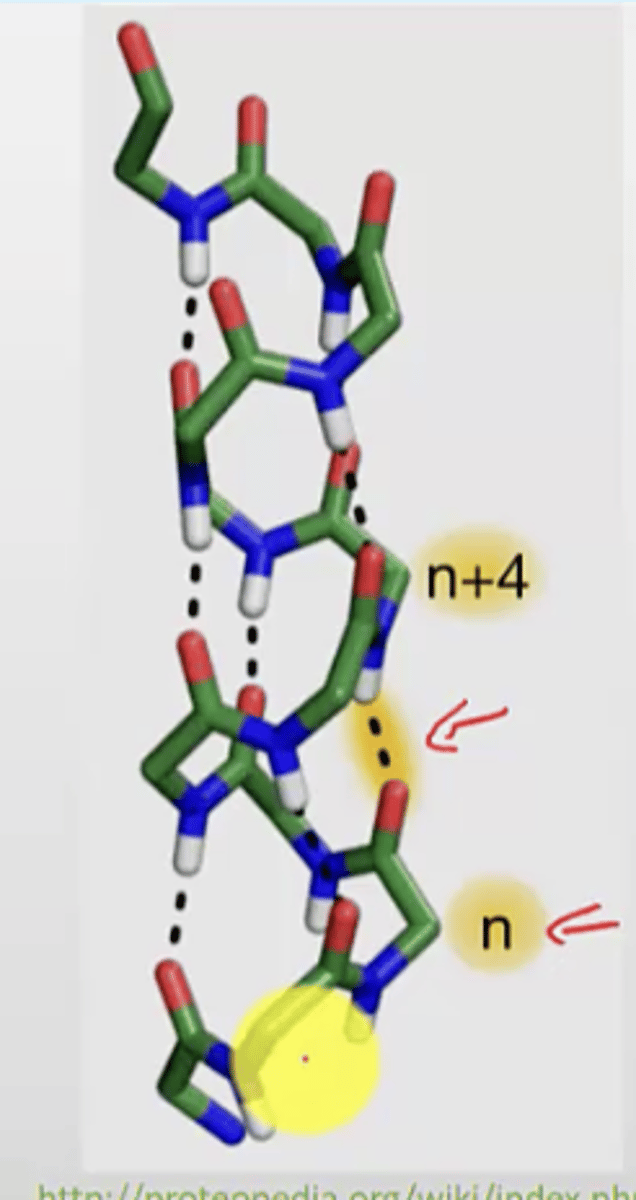
Relationship between residues and number of turns in a helix
3.6 residues per helical turn
- you can use this to calculate the length or number of turns of the helix
Distance along a helix axis for one turn is called
Pitch
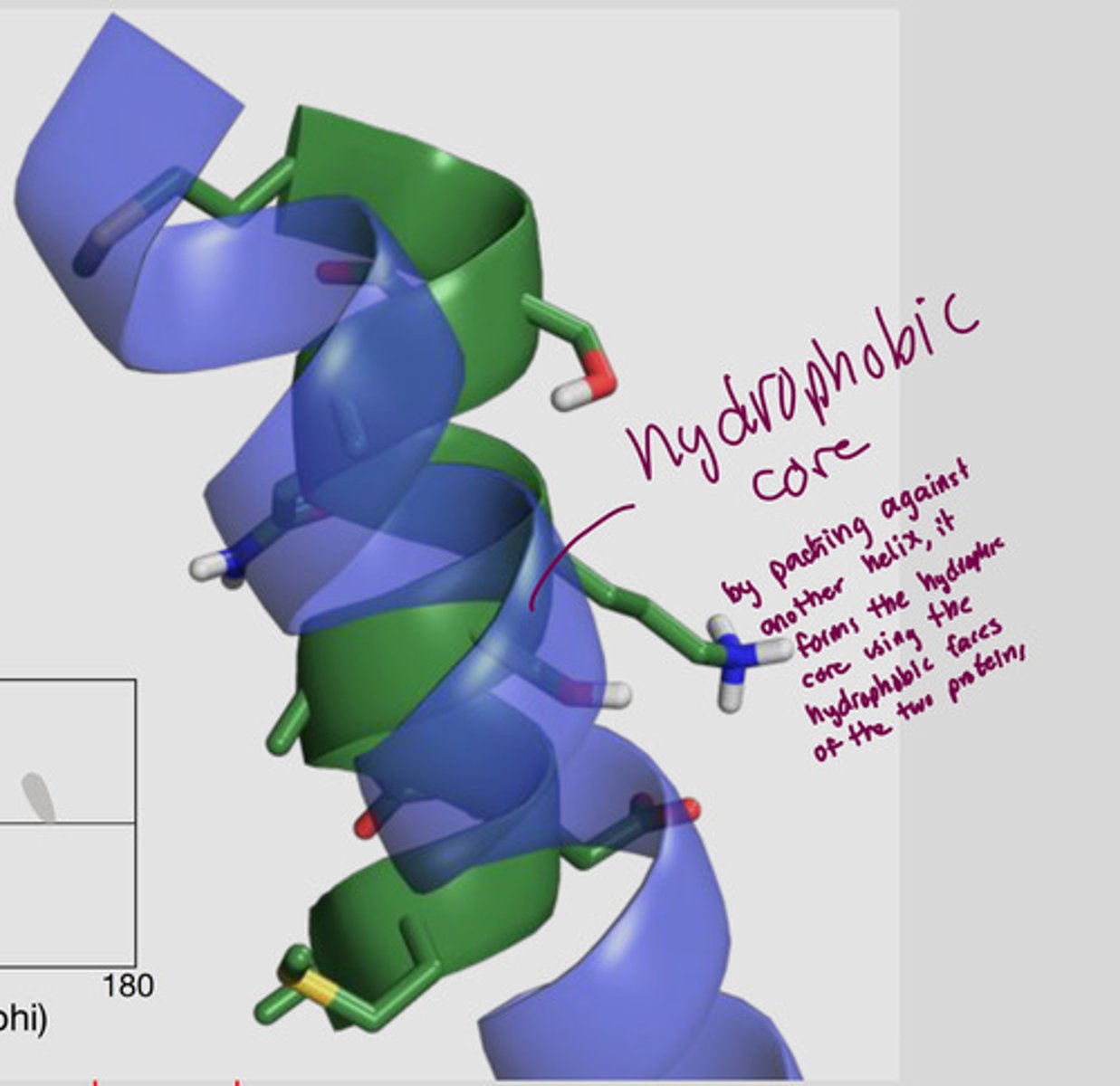
How does the alpha helix have faces?
R groups sticking out from the side have same properties (ex. one face has hydrophobic residues facing out, another side has hydrophilic residues)
all are about 3-4 residues apart --> forms hydrophobic core (forms when the hydrophobic faces of two proteins merge)
What is the most common angle region on the plot?
The 3rd quadrant, exactly where the right-handed alpha helix is
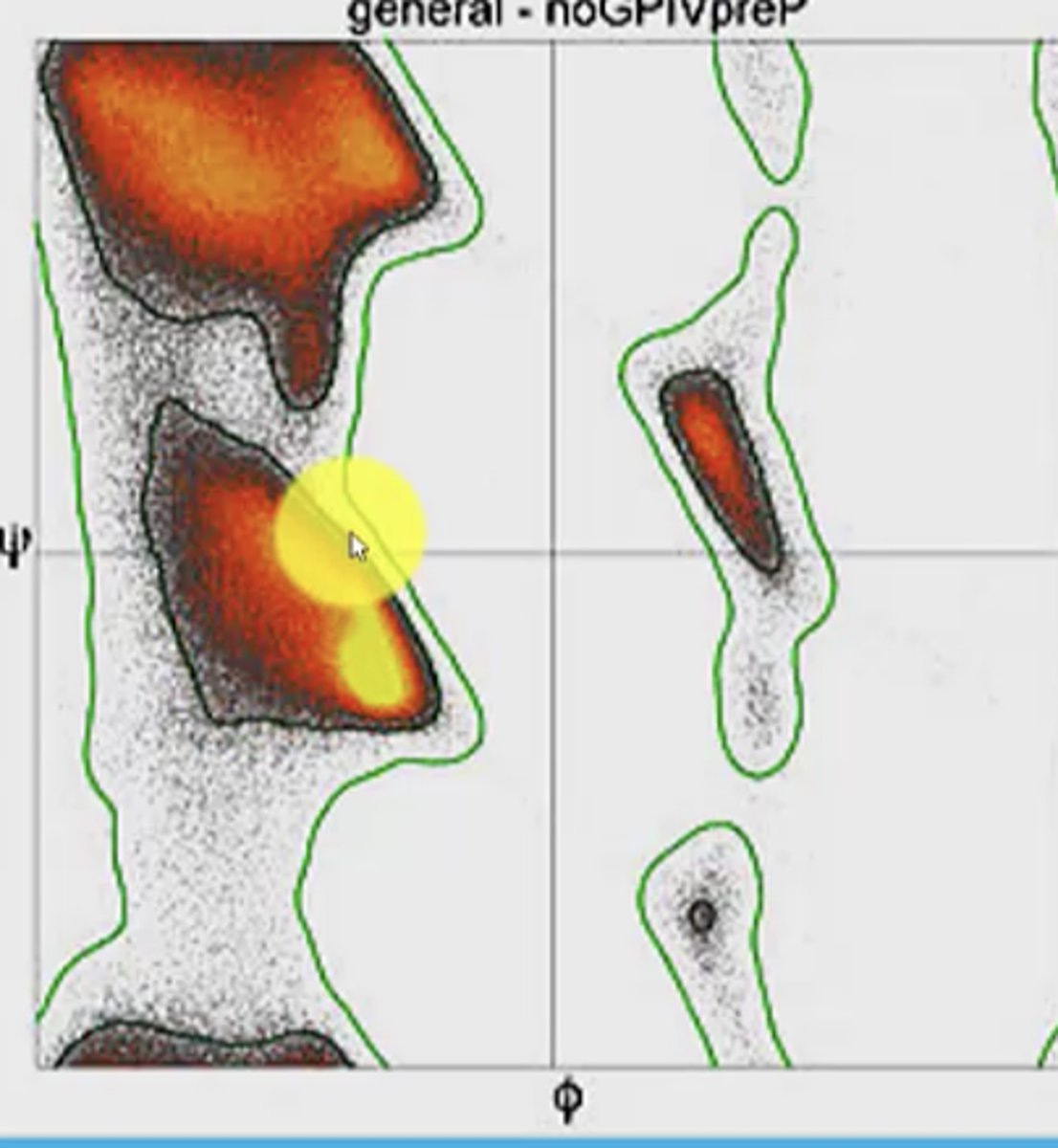
Why is the alpha helix more common?
>25% of all aa in proteins have alpha helix
Which amino acid is the most favourable to form alpha-helices?
Alanine (delta G is zero)
Which amino acids are known as the helix breakers? Why?
Proline and glycine - have a high, positive delta G (around +4)
Proline:
- cyclic
- too rigid, steric clashes
- helix would kink/bend
Glycine:
- small
- more conformational freedom (so it would favour the unfolded conformation over the helix)
What would happen if you had a long chain of alanine in your polypeptide at pH 7?
At pH 7, ala is negative
So, a long chain of only ala would repel one another and would not form the alpha helix
What determines the ability for amino acids to form the helix?
their properties, surrounding environment, and number of residues
Alpha helix has a large macroscopic helix dipole moment
an overall dipole moment due to the sum of the individual smaller dipoles of the carbonyl groups of the peptide bond pointing towards the helix axis
Carbonyl O = partially negative
Amide H = partially positive
All peptide bonds in the helix have similar orientation
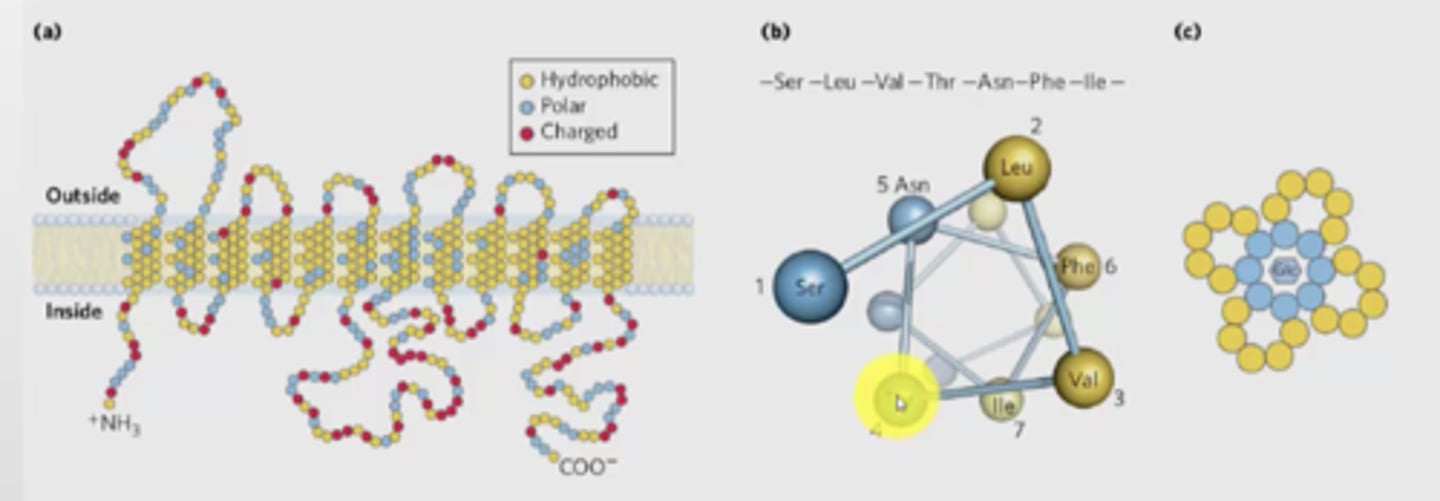
Alpha helix and the primary structure
By looking at the primary sequence, you can predict if that sequence can form an alpha helix
Look at this by making a helical wheel, looking down on it
If you number them based off the order from the top, you can see which amino acids in the primary sequence would form interactions
Example is this pic: 1 and 4 forms interactions since it is a + and -
2, 3, 6 are all on one face of the helix since they are all non-polar (creates the hydrophobic effect)

What are critical factors of alpha helix?
1. Intrinsic properties of amino acid residue to form alpha helix
2. interactions between R groups (especially those spaced 3-4 residues apart)
3. bulkiness of adjacent R groups
4. occurrence of Pro and Gly residues
5. interactions between amino acid residues at the ends of the helical segment (electric dipole)
Function of amphipathic alpha helix
the alpha helices are closely beside other structures within the protein
so, the exposed faces of the amphipathic helix is important for what surrounds the helix
How can alpha helix traverse membranes?
transmembrane helices are represented as angled rows of 3-4 amino acid residues, each row shows 1 turn of the helix
(c) shows how the helix is arranged in the membrane, shows that each amphipathic - produced a channel of charged molecule that are able to interact with glucose (hydrophilic portions are buried inside)
Why is proline a helice breaker
lacks NH group and has a ring structure that prevents it from having a phi value to fit into a helix
no H on amide group
- even if proline is in trans or cis position, the steric clash will always happen
Why is polypeptide II helix (collagen) important?
the sequence is PPGPPGPPG
- unusual structure
- forms left-handed helix
- not stabilized by H bonds
- 1/3 of residues in the secondary structure are proline
- however, very important in body
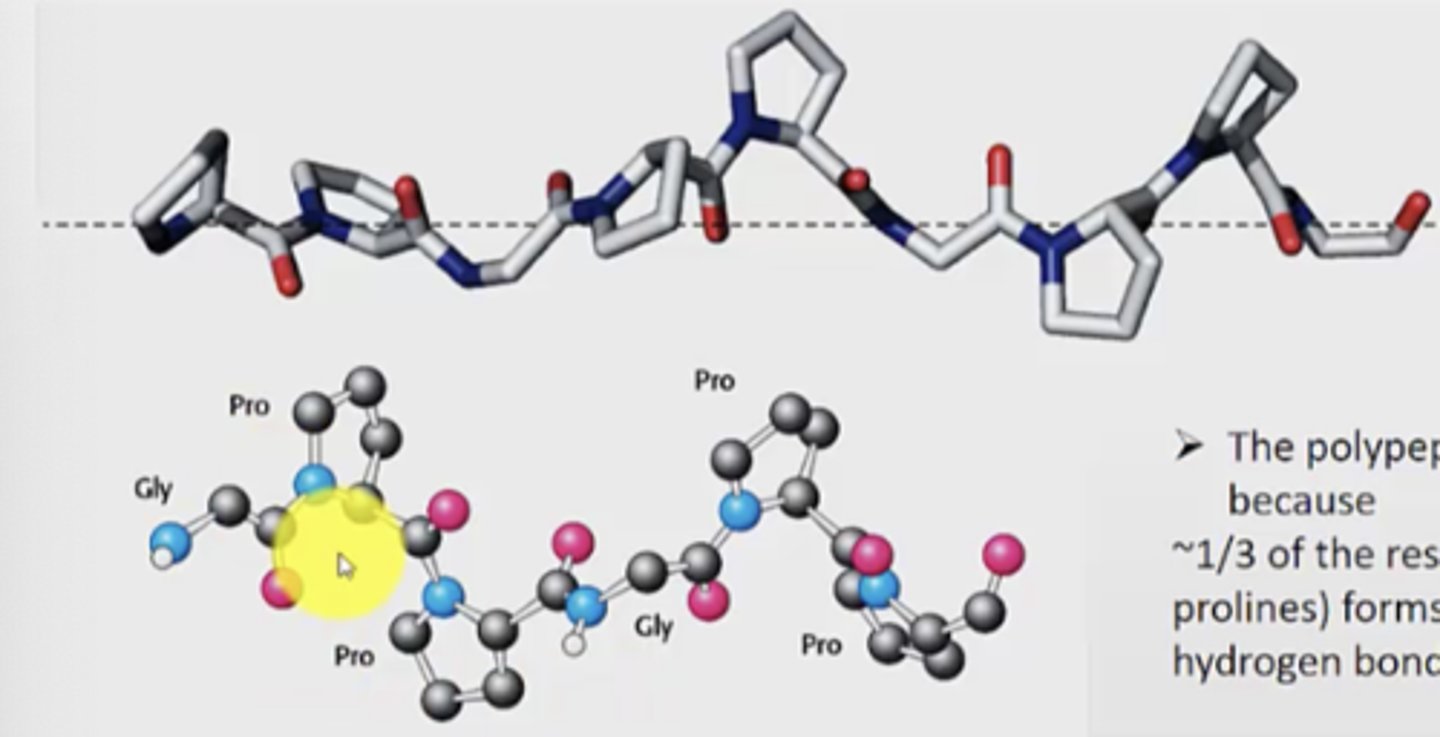
Describe the structure of polypeptide II helix in a cross section
- each strand of collagen in a cross section forms a hydrogen bond to the other 2 strands
- every third residue must be glycine because there is no space in the centre of the helix
- the ring is on the outside

Functional consequences of collagen misfolding/mutation/deficiency
1. Osteogenesis Imperfecta
- known as brittle bone disease
- inherited bone disorder that is present at birth
- soft/brittle bones not formed properly
2. Scurvy
- lacking vitamin C
- vitamin C is needed to product mature collagen, so less vitamin C means less collagen
Conformation and location on plot for beta-sheets
- located in the top 2nd quadrant
- has an extended backbone conformation (unlike helix where it is coiled with turns)
- goes from N->C terminus
- H bonds between the amino and carbonyl groups of adjacent strands forms the beta-sheets
Where is the b-sheet more
edge of b-sheet, side chains extend above and below the plane
- alternating pattern in amino acid resides
- so the sheet can have two sides
What angle is the backbone of b-sheet more extended? How does this form the beta-pleated sheet structure?
backbone is more extended with the ψ (psi) dihedral (N-Ca-C-N) in range of 90deg-ψ-180deg
the planarity of the peptide bond and tetrahedral geometry of the Ca creates the pleated structure
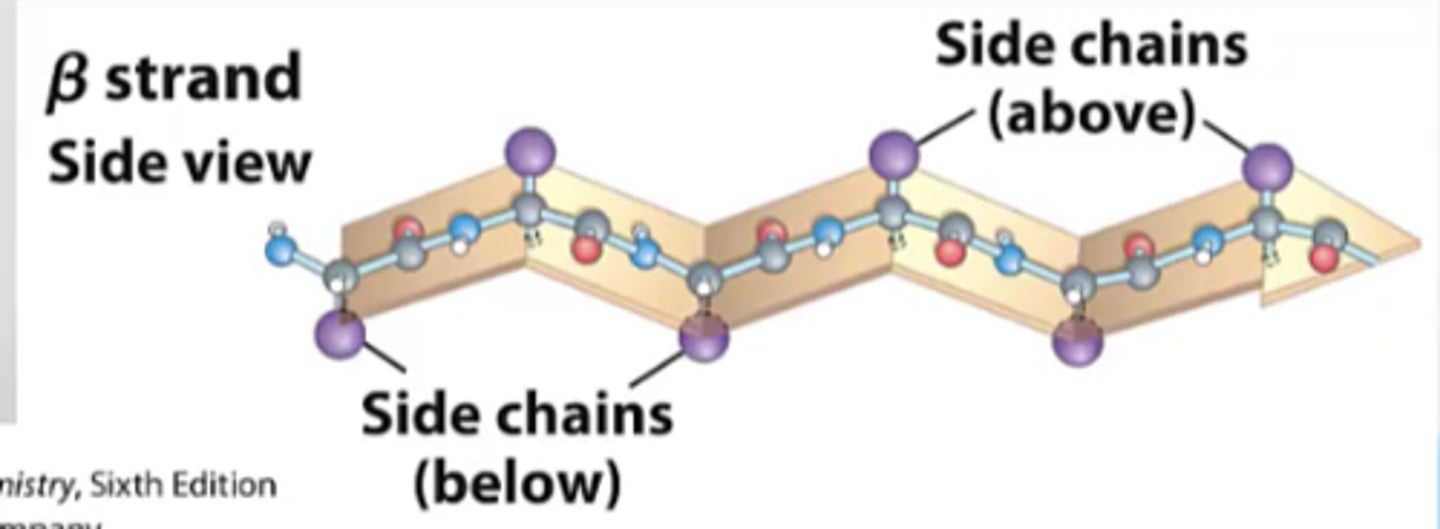
What is the sheet arrangement held together by?
H bonds between more distal backbone amides
Why do pleated sheets have up down direction?
since side chains protrude from the sheet in alternating up and down directoins
Two types of beta-sheets
1. MD2 LPS binding protein
- mostly anti-parallel sheets
2. CsTII
- mostly parallel sheets
Antiparallel strands vs. parallel strands
1. anti-parallel
- H bonds are straight
- connected to each other via loops
2. parallel
- H bonds are bent slightly
- strands taken from different parts of the protein, not connected at a point
So, B-sheets contribute to protein stability over long distances, since the run long through the protein structure
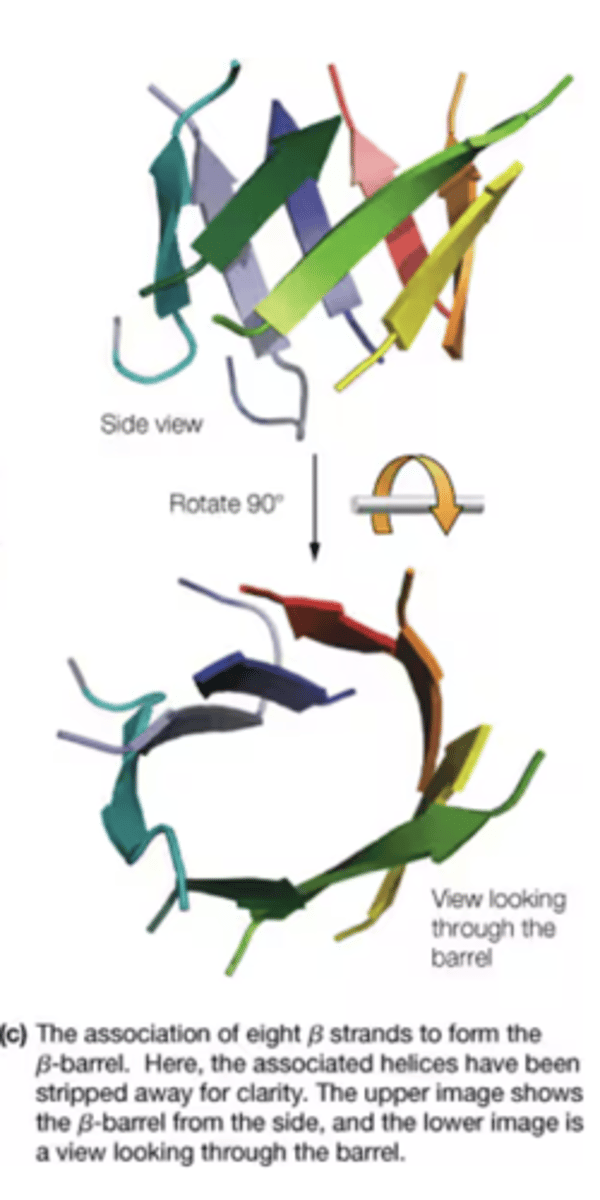
How is a B-barrel formed
A contiguous B-sheets form a barrel (a common domain fold)
- side chains are exposed to hydrophobic core
What does the bottom blue region correspond to?
turn and loop regions - because
BUT, for loops any allowable phi-psi angles are ok, so you can find loop residues in all three quadrants of the plot
What are a-helices and b-sheets connected by?
loops
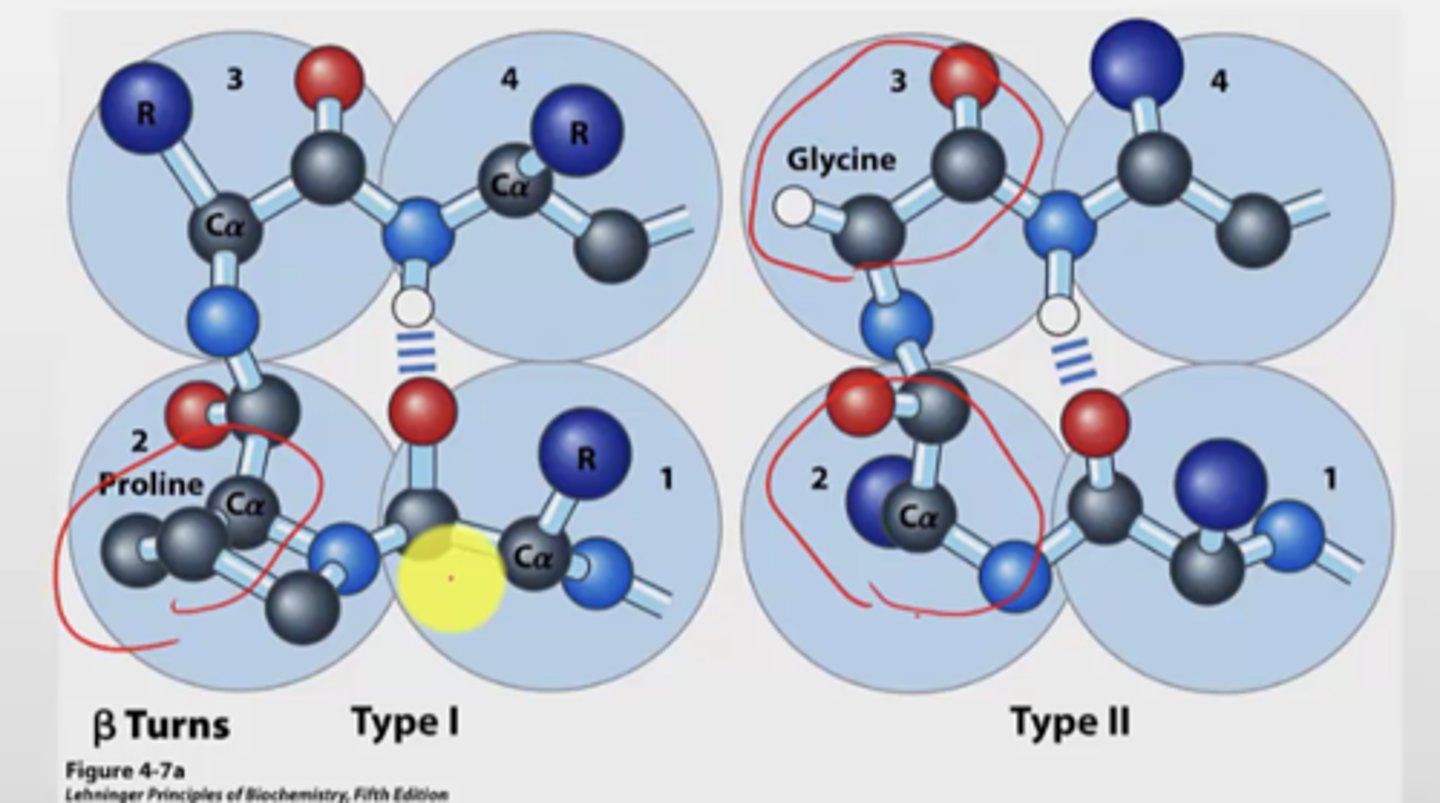
B turns
- occur when strands in b-sheets change their direction
- turns are very sharp, 180 deg over 4 amino acids
- turn is stabilized by a hydrogen bond from a carbonyl oxygen to amide proton three residues down the sequence
- proline in position 2 or glycine in position 3 are common
- connect helices, sheet strands
B turns type 1 vs type 2
Type 1:
- more common (2x more frequently)
- proline most common in position 2
Type 2:
- glycine at position 3
- proline is also most common at position 2 here
Where are amino acids with R groups found on the plot?
For large R groups, the allowable angles and regions on the plot they can occur in are smaller
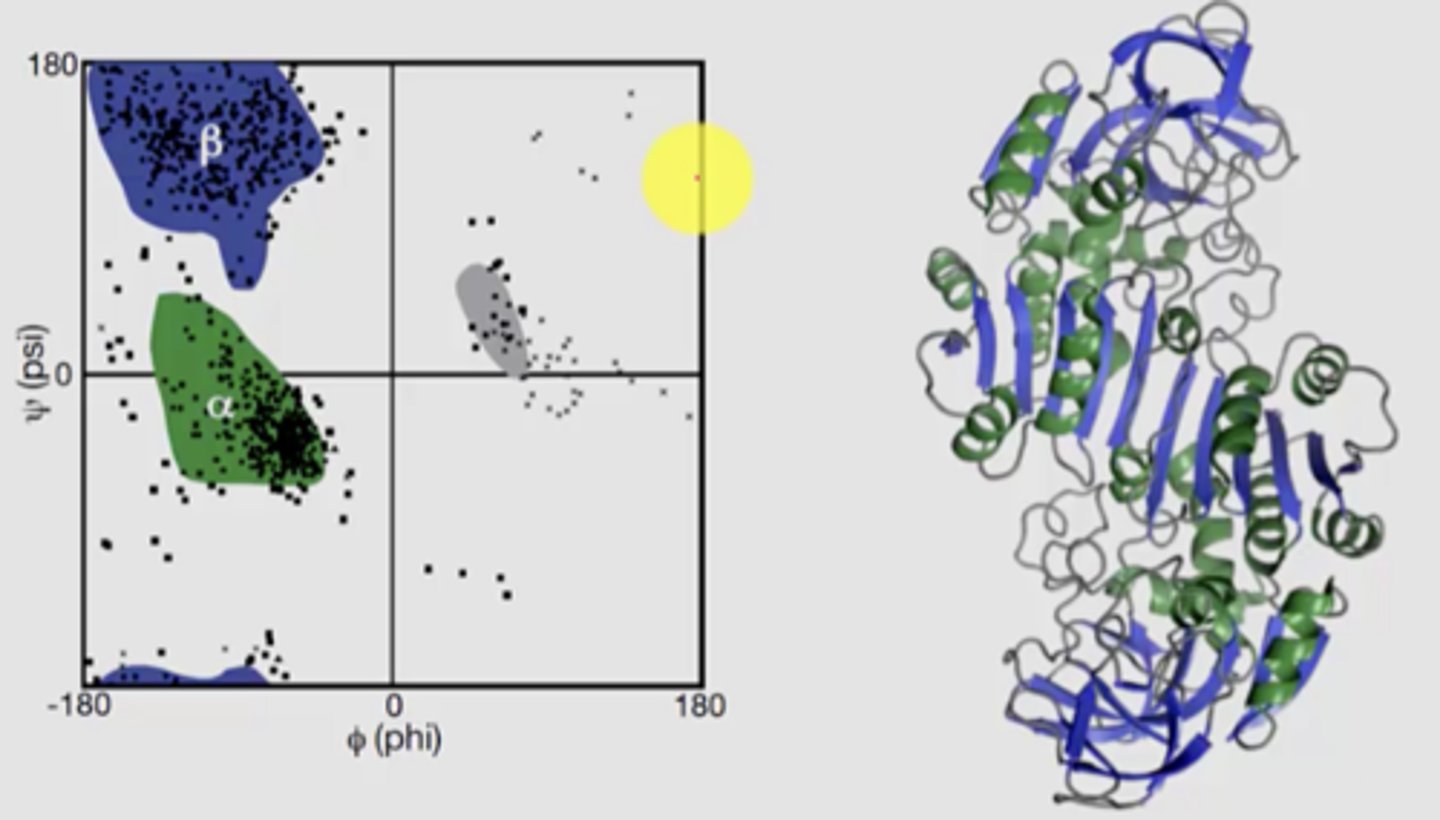
Ramachandran plot for alcohol dehydrogenase
- has a prosthetic group
- contains mixture of a-helixes, b-sheets, and loops
- residues (black dots on plot) are distributed over all regions of the plot
Two major classes of tertiary structures
1. Fibrous proteins
- insoluble
- made from single secondary structure
- mostly structural role, major components of skin and CT
2. Globular
- water soluble
- lipid soluble
depends on location of cell
Fibrous protein examples and definition
elongated molecules with well defined secondary structures
- keratin (hair, fingernails, feathers, scales, intermediate filaments)
- fibroin (silk cocoons)
- collagen (CT, matrix in bone)
alpha-keratin
- double helix that coils around itself
- large hydrophobic residues repeat every 4 positions
- 3.6 residues/turn
- each helix has a hydrophobic side (which is how the helix coils around itself, since the hydrophobic sides are the interface between two long helices in the coiled-coil structure)
What type of secondary structure is silk fibroin? What is the structure of silk fibroin?
Where does strength and elasticity come from?
- B-sheet, tightly packed together
- has alternating layers of alanine and glycine
- fibers are very flexible because bonding between sheets involves interactions between the side chains
- strength of silk comes from the alternating directions of the b-sheets along the length of the fibers
- elasticity comes from compact folded regions which interrupt the close-packed b-sheets
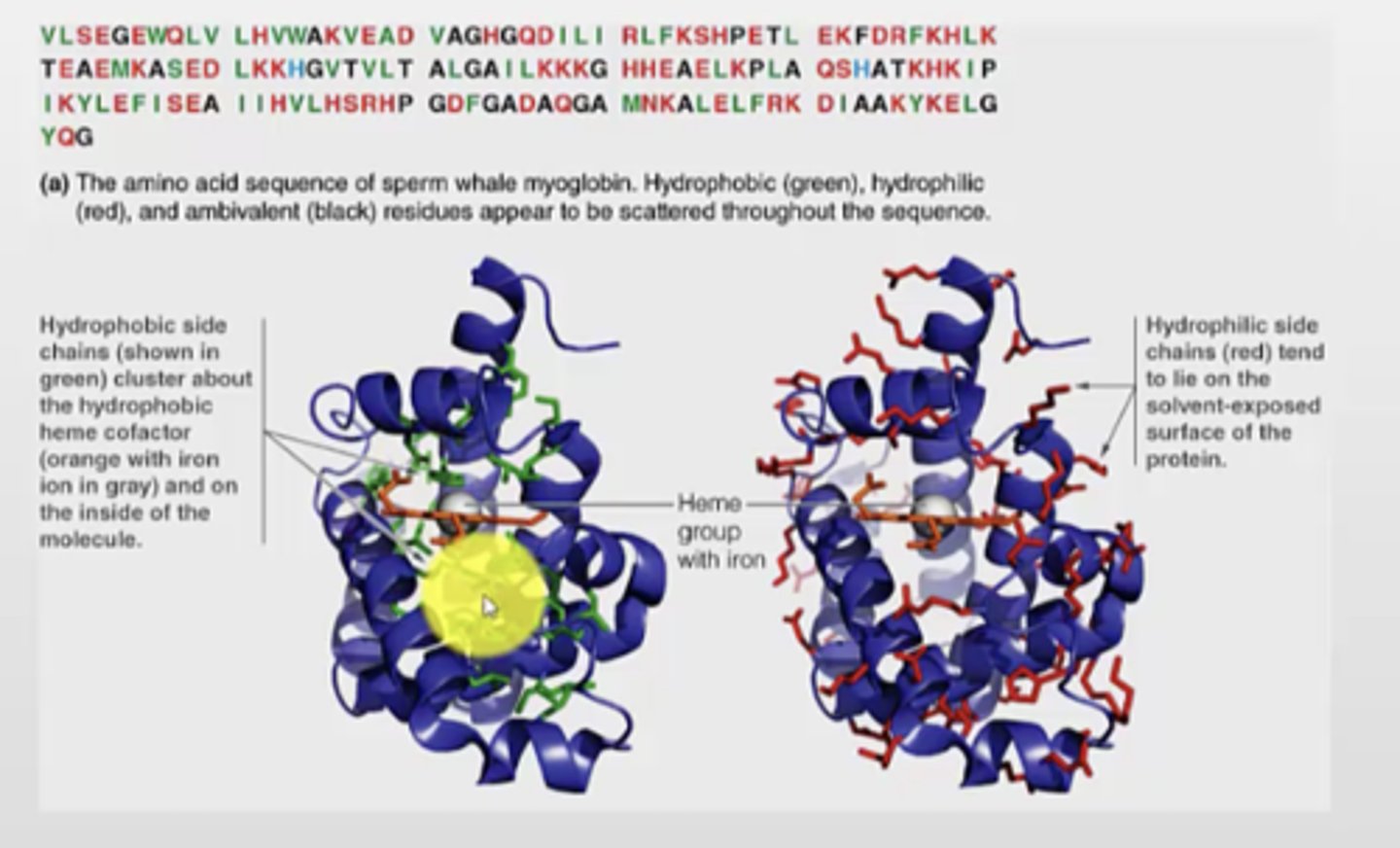
Why is distribution of hydrophobic and hydrophilic residues important?
Use myoglobin as an example
- burying hydrophobic residues is important for protein folding
- hydrophobic core
- from the primary sequence, you cannot see how the hydrophilic and phobic side chains are distributed
ex.
- hydrophobic side chains cluster in the hydrophobic heme cofactor in the middle
- hydrophilic side chains lie on the solvent-exposed surface
Globular proteins structure
- diverse structures, varying amounts of helix, sheet, and loops
- larger proteins have 2+ domains of compact folded structure
- typical domain is 200 amino acids and fold independently
Motifs
- like a subcategory of domains / super secondary structure
- domains can be made of several motifs
- motifs can be simple or complex
stable arrangement of two or more secondary structure elements and their connecting loops
Why do we arrange globular proteins into motifs and domains?
Difference between motifs and domains
- to make sense of globular structures and break it down into smaller bits
Motifs:
stable arrangement of two or more secondary structure elements and their connecting loops
Domains:
part of a polypeptide chain that is independently stable, has a particular, independent function from the rest of the polypeptide
Domains can have motifs
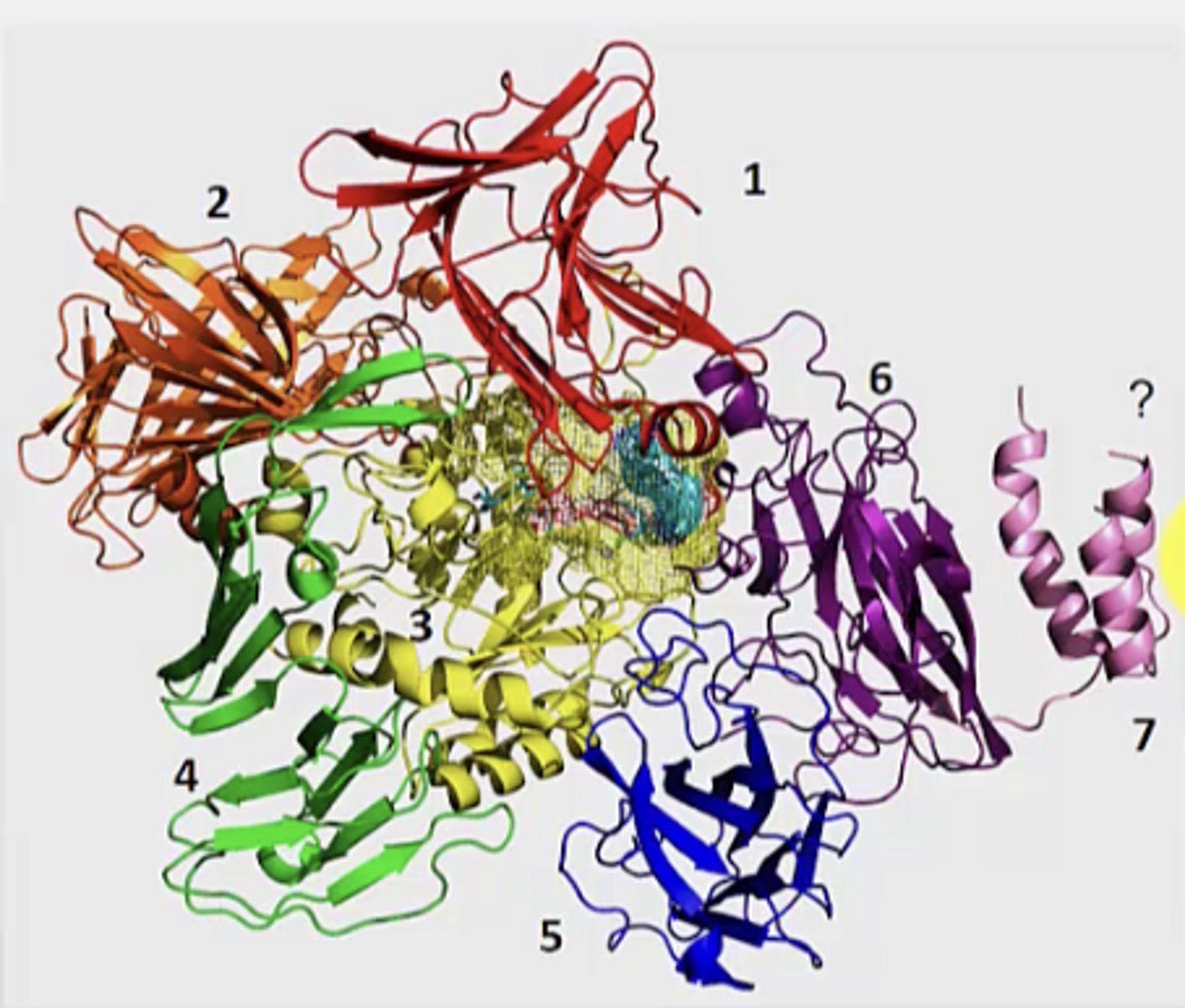
What are the 7 domains of streptococcus glycoprotein hydrolase? What is this an example of?
Example of multi-domain protein
1. B-sandwhich
2. supersandwich
3. b/a barrel
4. b-sheet
5. jelly roll (blue)
6. jelly roll (purple)
7. helical bundle
Tertiary vs quaternary structures
1. tertiary
arrangement of secondary structures within a single polypeptide
2. quaternary structure
- arrangement of several polypeptides within a protein complex
- formed by assembly of individual polypeptides into a larger functional cluster
Where are the active sites of enzymes found
in loop regions or between two domains
2 examples of quaternary structure
1. antibody - IgG
2. growth factor receptor
3. ATPase
What holds tertiary and quaternary structures together
1. hydrophobic effect
- increases entropy of surrounding water
- release of water from the solvation layer around the molecule
2. hydrogen bonds
- N-H and C=O of peptide bond between polar R groups
3. van der walls
- medium range weak interactions, can contribute to the stability in interior of the protein
- between all atoms
4. electrostatic interactions
- long-range, strong interactions
- between charged groups
5. covalent bonds
- ex. disulphide interactions
Are protein folds reused or made newly in new proteins? Why is this important?
they are reused instead of using a completely new one
- number of protein structures keep increasing
- but number of folds that exist become constant
- therefore, combination of folds has many different possibilities, which is why they are reused
Domains are modular
What environment do proteins fold in? Why is this important?
Aqueous environment
NOT VACUOLE
Important since the aqueous environment allows the proteins to achieve their native conformation
How does protein folding reduce entropy
When unfolded, the protein has many conformations it can be in
Once it folds, it achieves one native conformation
This plays AGAINST protein folding since it decreases entropy
Why is the aqueous environment important for protein folding?
hint: decrease of entropy water, hydrophobic effect
Free water molecules have high entropy (free rotation, can bind with many molecules)
When water is on the hydrophobic surface of a protein, it will have fewer allowed conformations (decreased entropy)
This is important since the hydrophobic effect drives protein folding since it buries hydrophobic residues in the middle.
Why is the hydrophobic effect cooperative in protein folding? What does cooperativity mean?
Because it will gather all the hydrophobic residues in the middle efficiently (once first hydrophobic molecules gets folded in the inside, the other phobic residues will be more likely to interact with the middle, forming the hydrophobic center)
Cooperativity is when one molecule undergoes a change that drives other molecules to undergo the same change fast
What are the final contributor to protein folding
Non-covalent interactions, salt bridges, van der waals
Contribute to a favourable and net decrease in enthalpy of the protein as it folds
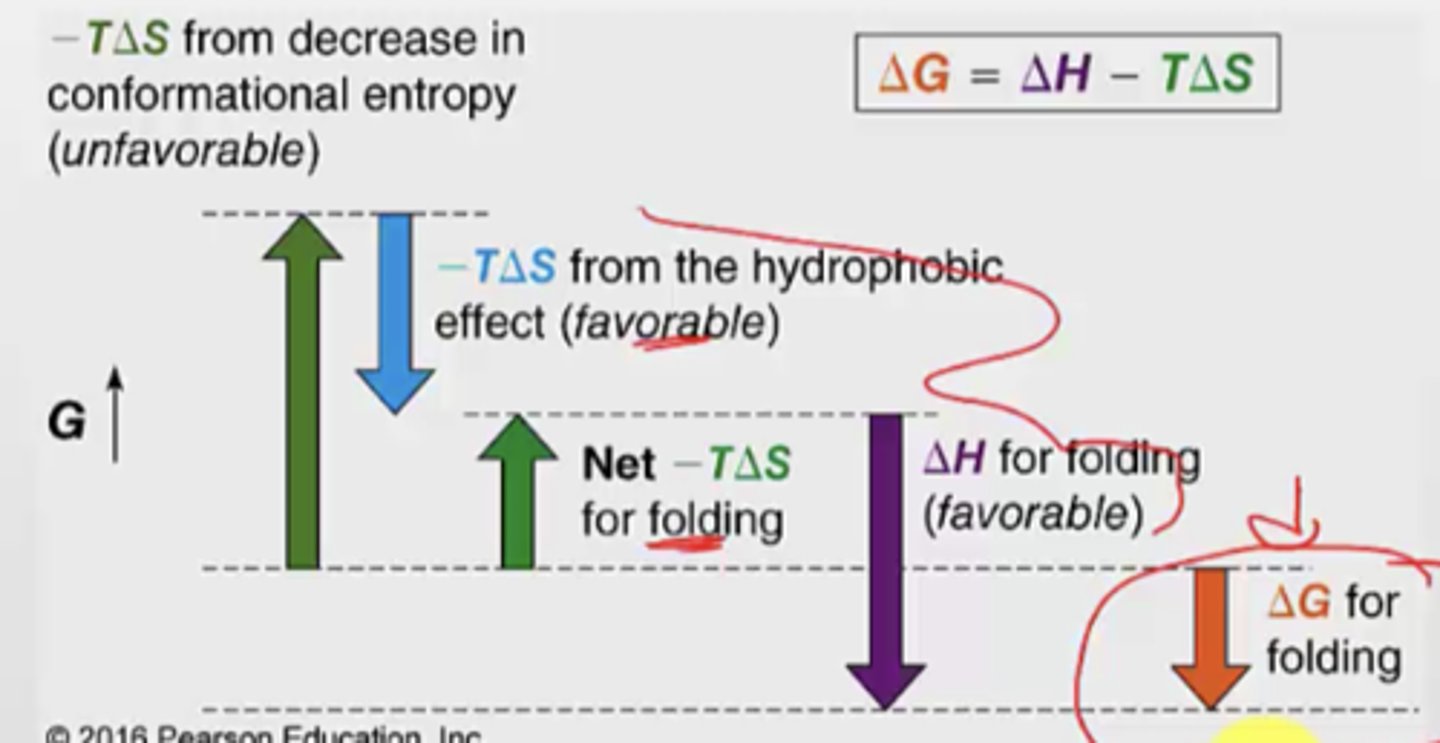
What are the 3 thermodynamic factors that influence folding and stability of proteins?
1. Favourable intramolecular enthalpic interactions (-H, -G)
- charge-charge interactions, hydrogen bonds, van der waals
2. Unfavourable loss of conformational entropy (-S, +G)
- Since folded state has less conformations and less entropy
3. Favourable gain of solvent entropy from burying hydrophobic groups (+S, -G)
- the hydrophobic effect
Is protein folding favourable or unfavourable? Why?
Favourable
the conformational entropy of chain is very positive G -> this only happens in unfolded
SO, since folding doesn't have that, it is more negative G
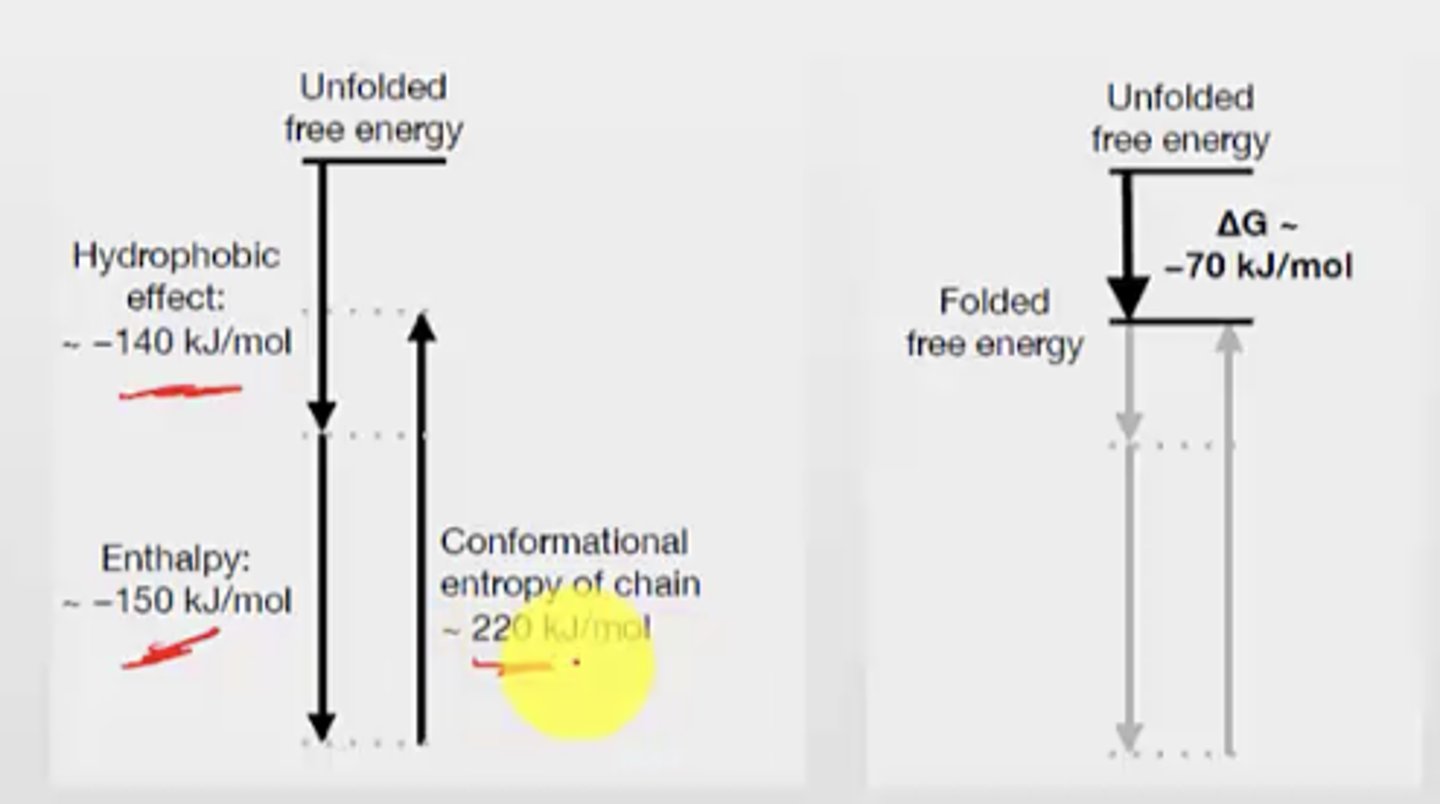
What makes protein folding favourable?
Different enthalpic factors can contribute to the -G for protein folding
What are 3 ways to denature proteins
1. heat to melting point
2. change in pH
3. add chaotrope (like urea or ethanol)
- these are very polar, so when present in very high concentrations is will alter the hydrogen bonding of water which will decrease impact of the hydrophobic effect (which is a big favourable component)
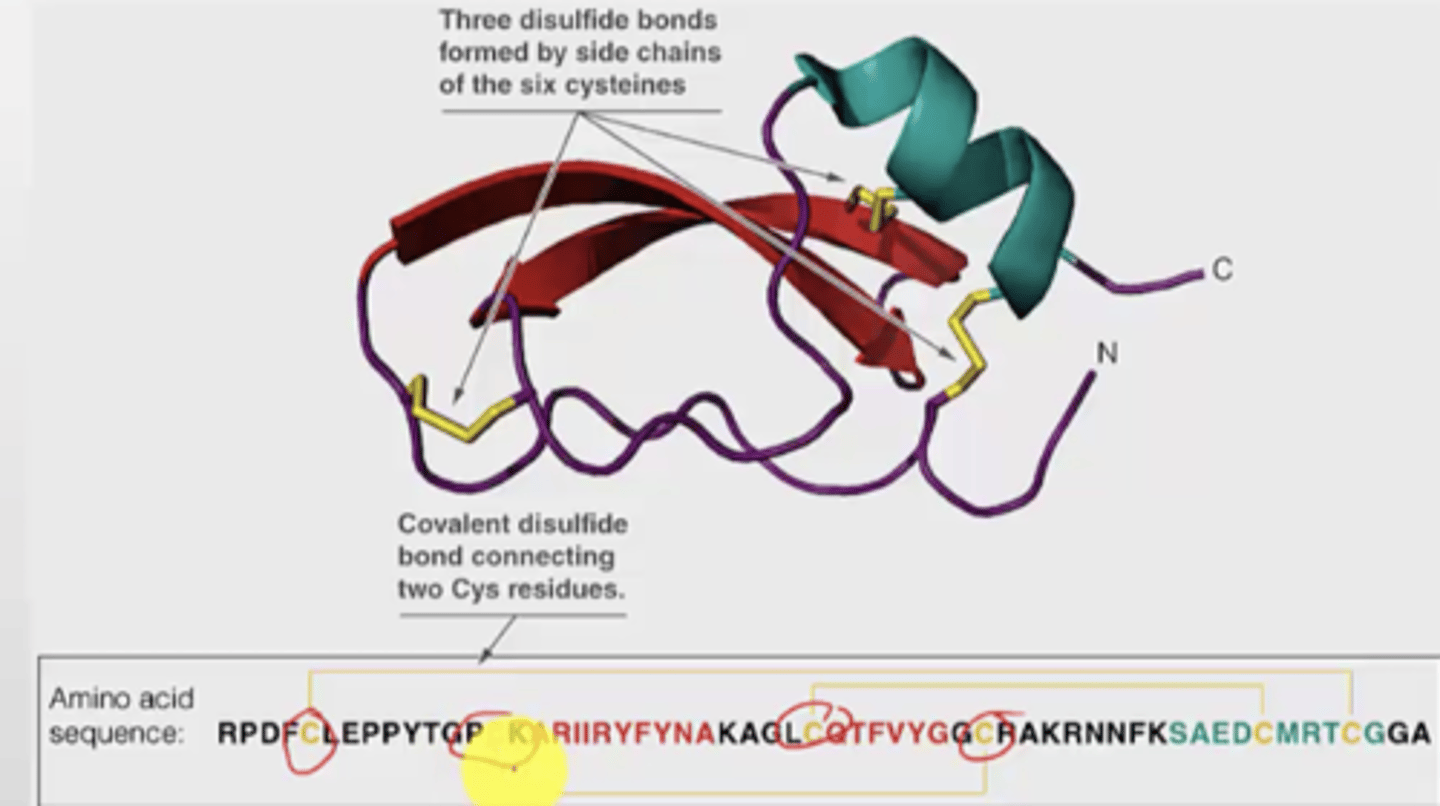
Why are disulfide bonds important?
It links secondary structures elements together
They increase stability
Example: BTI3D structure
- has three disulfide bonds (covalent) by connecting two cysteines
- the cysteine molecules used for bonding is also important (cannot be random)
- even if you cleave just one of the disulfide bonds, it will impact the structure A LOTT
How can Zn2+ be a stabilizing factor?
You can have a zinc finger domain bound to a Zn2+ ion
- two histidines and two cysteines bind to a Zn2+ ion - stabilizes this section
- common in DNA-binding proteins
Which prosthetic group can be a stabilizing factor? Example with myoglobin.
Apomyoglobin has no heme group
When a heme group binds, it stabilizes the folded myoglobin structure and gives it a red colour --> producing holomyoglobin
What is NMR, why is it important?
NMR used to determine tertiary structure
Exposes protein solutions to magnetic field
Anfinsen Experiment - Intro
- Proof that primary sequence helps determine tertiary structure
- used RNAse A (ribonuclease, so it is easy to read)
- Urea (chaotrope) and mercaptoethanol together dentatures the ribonuclease
- Removing denaturants (urea), protein refolds, correct disulfide bonds reform (after refolding) and protein regains activity
From the Anfisen experiment, what determines native conformation
the sequence of protein
Conclusion and procedure of Anfisen experiment
1.
added urea and B-mercaptoethanol to RNAse A, removed them, allowed it to reform -> 90% activity restored
2.
Control experiment:
- denatured and reduced the disulfide bonds
- before dialyzing, he adds an oxidizing agent to neutralize the mercaptoethanol to reform disulfide bonds
- THEN the protein reformed
So, he allowed the disulfide bonds to form while protein was denatured --> 1% activity restored
3.
Adding to part 2. he took the RNAse with mismatched disulfide bonds and added small amount of mercaptoethanol
- this allowed the incorrect disulfide bonds to the break and reform into the right bonds --> 90% activity restores (HAPPENED WITHOUT UREA PRESENT)
General conclusion about the Anifsen experiment
RNAse protein could not reach its native form when the disulfide bonds were allowed to form randomly with urea present
When urea and reducing agent was removed, the native form as achieved
So, first protein folds, then disulfide bonds form
FINAL conclusion about Anifsen experiment
1. most stable thermodynamic conformation is the native form
2. primary structure is enough to dictate the tertiary structure (3D folds)
Can Ai be used to predict structures?
yes
Levinthal's paradox
It is mathematically impossible for protein folding to occur by randomly trying every conformation until the lowest-energy one is found
If protein samples all possible conformations, rejecting each until it reaches the most stable conformation, folding would take 1027 years
So, not all states are samples (since folding occurs in seconds), instead there are intermediate states with some native secondary structures already existing
Width
number of conformations
Is the native state high or low energy?
Lowest energy state possible
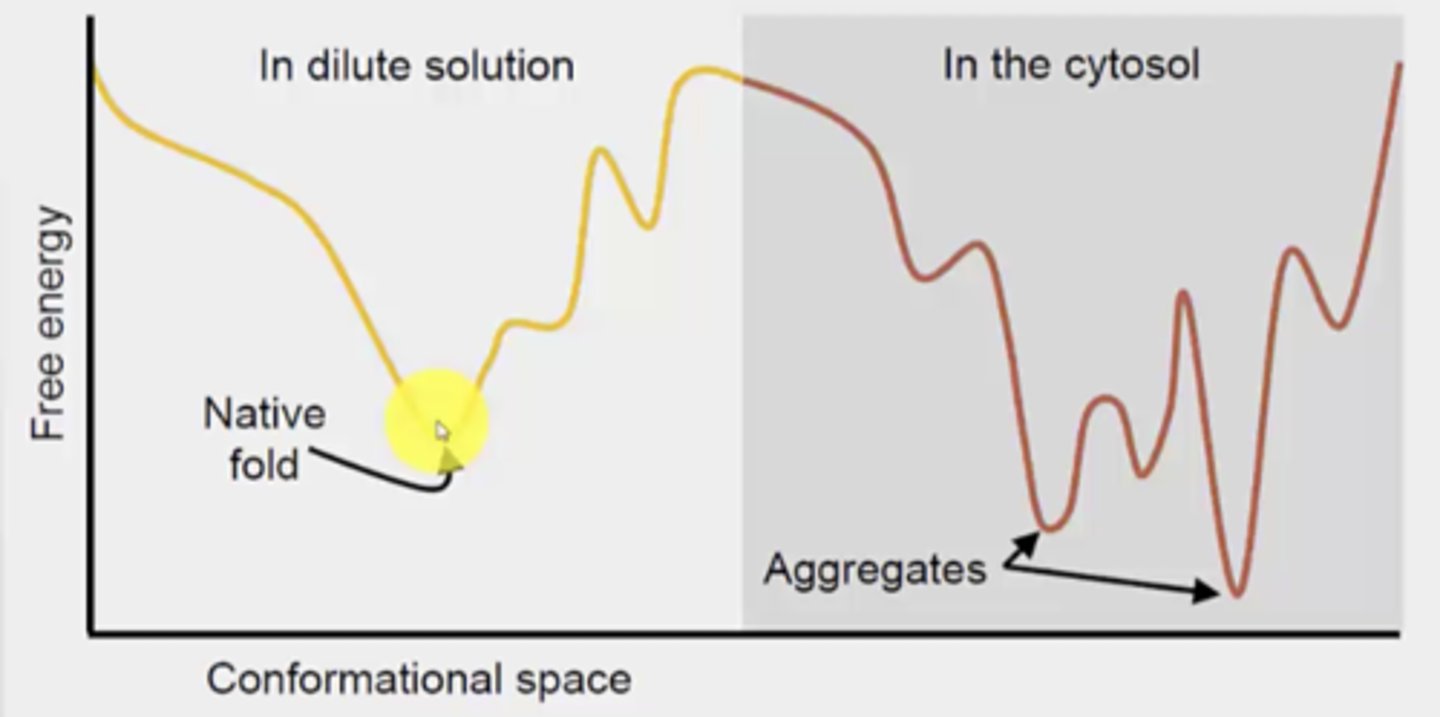
Why is protein folding different in the cytosol vs. aqueous (something else is present in the cytosol that impacts it)
presence of proteins and RNA in the cytosol (can interfere with folding)
What is the risk of folding in the cytosol
aggregation can happen
the aggregates have lower free energy than the native fold, so the aggregation form would dominate if it were to occur
Chaperons + example
Promote correct folding (avoids aggregation)
ex. protein disulfide isomerase to correct formation of non-native disulfide bonds
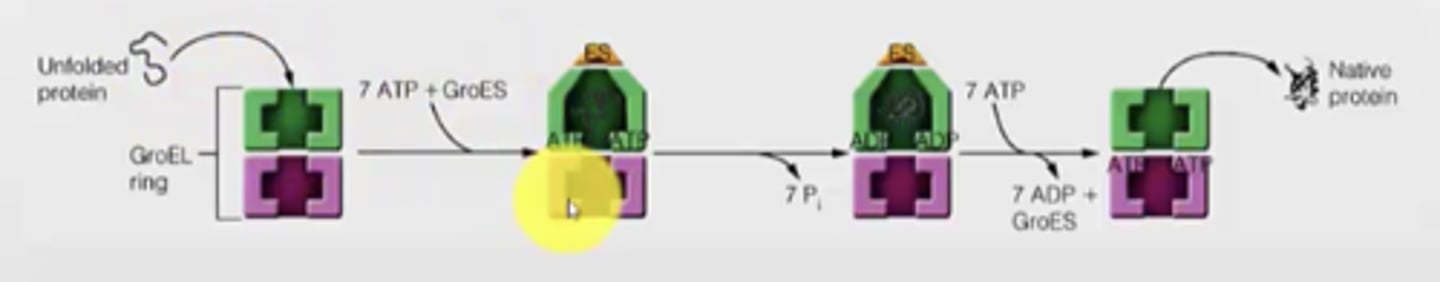
How does a chaperon work?
Example with GroEL ring for E. coli --> helps prevent hydrophobic interactions that would lead to aggregation
- chaperon is lined with hydrophobic groups to attract the misfolded protein
- Unfolded protein enters the chaperon
- ATP and GroES binds, causes a conformation change and interior now presents a hydrophilic group to promote proper folding
- now, properly folded protein can leave the GroES
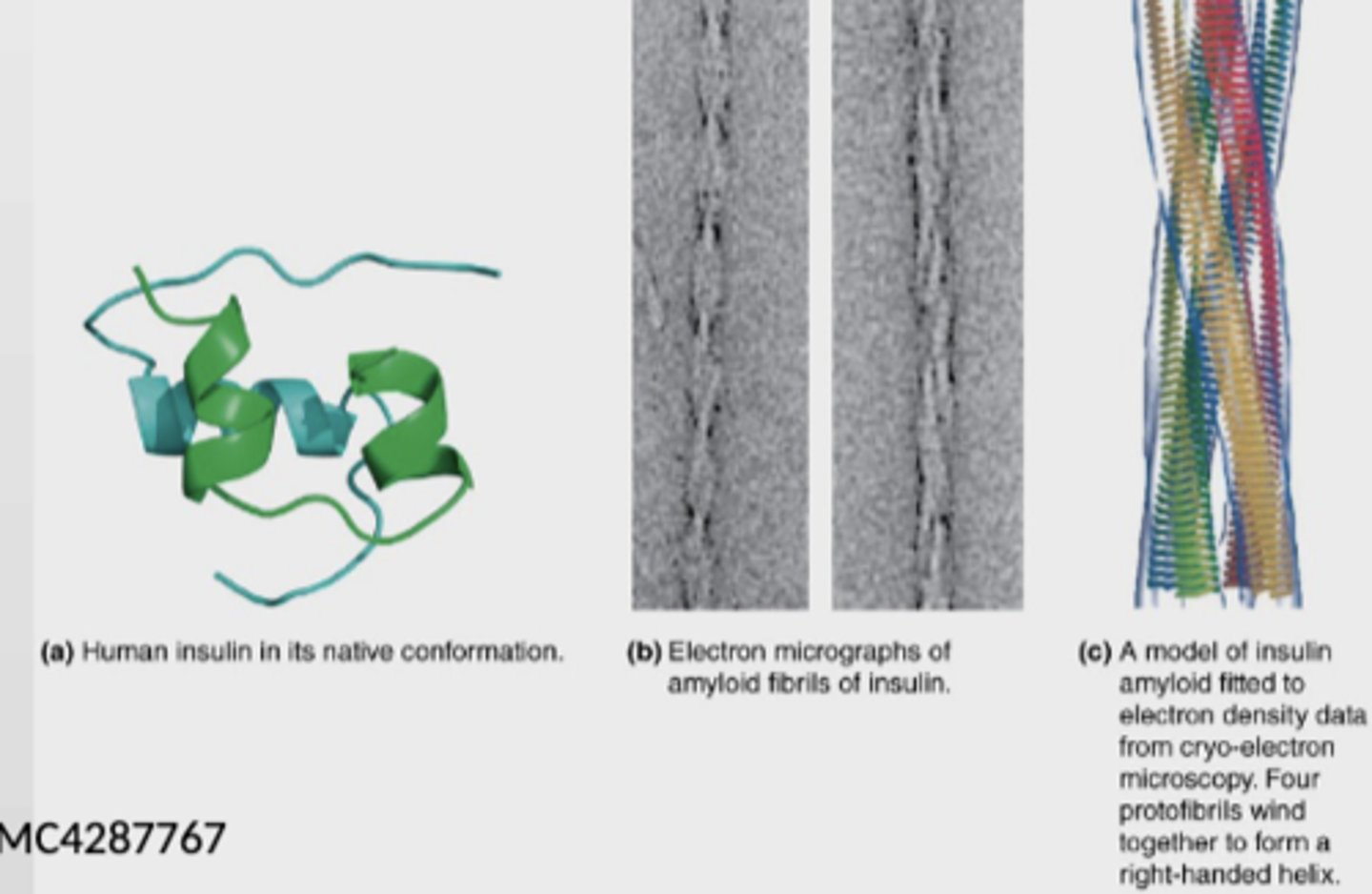
Amyloidosis - disease caused by misfolding
disease caused when normally soluble proteins become unfolded, secreted from cells and forms insoluble protein structure
- unfolded proteins become entangled forming an amyloid fiber (protein deposit or aggregate)
- amyloid fibers have a high degree of b-sheets
- normal folding of protein is mostly a-helix
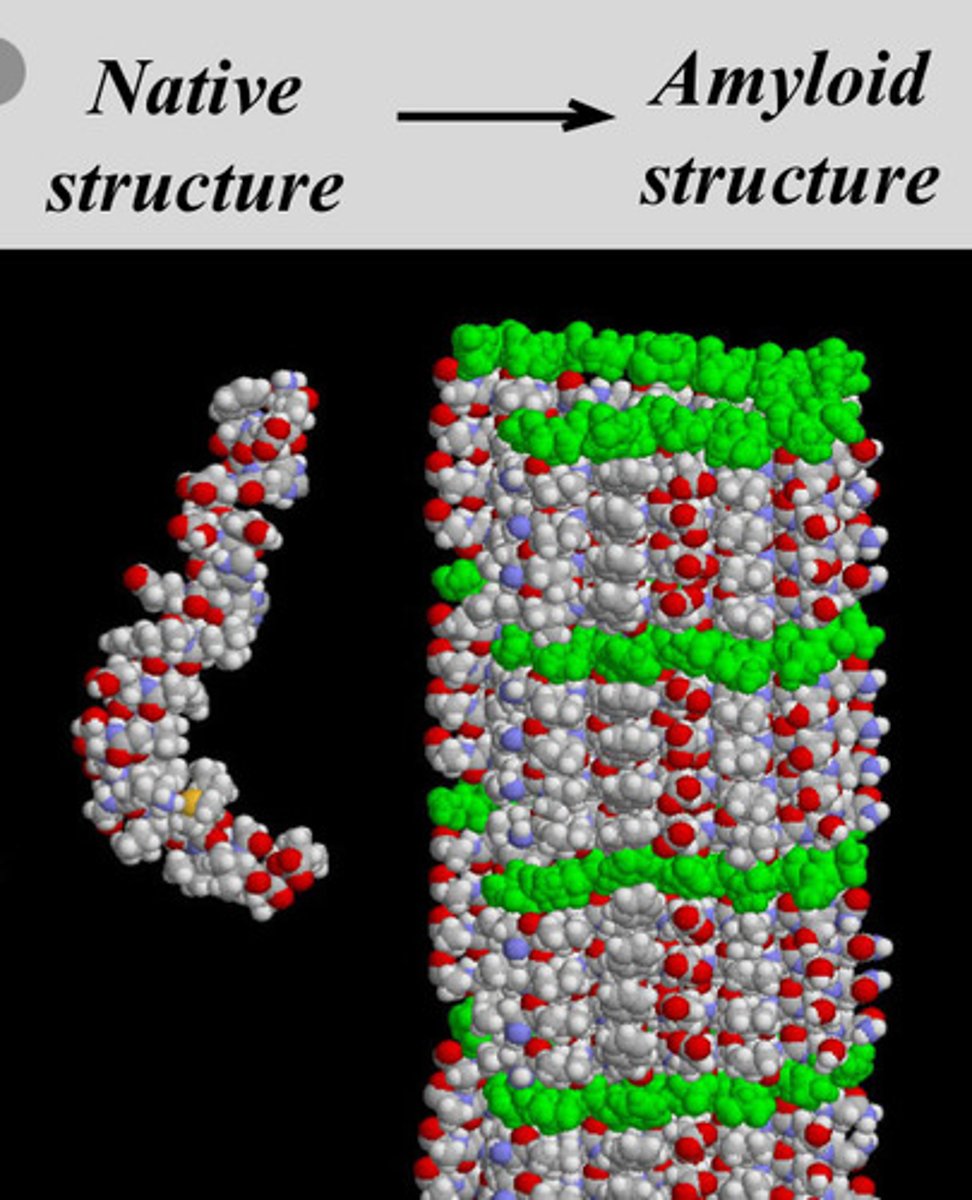
Insulin-derived amyloidosis - disease caused by misfolding
- rare
- causes poor glycemic control and increased insulin dose requirements since insulin absorption is impaired
Prions - disease caused by misfolding
proteinaceous infections agents (NO NUCLEIC ACIDS) which are structually different from the normal protein (PrP)
ex.
- Mad cow disease
- scrapie
- vCJD
When PrPc (normal) comes in contact with a prion, it is converted into PrPsc (misfolded infectious protein)
Principle of absorption spectroscopy (UV-Vis)
- molecules are excited by energy of different wavelengths from the electromagnetic spectrum
- when excited, molecules are in a higher energy state compared to their ground state (absorption)
- most useful for proteins/nucleic acids
- High energy transitions = absorption
What molecules absorbs UV/Vis region?
- Ring structures
- Highly conjugated molecules
- all the nucleic acid bases, W (tryptophan) and Y (tyrosine)
What is Beer's law
states:
- molar absorptivity is constant
- the absorbance is proportional to concentration for a given substance dissolved in a given solute
- measured at a given wavelength
- each protein measured with this has an extinction coefficient (how well they absorb)
What is fluorescence
energy released as photons after excitation as a way to return back to ground state
W and Y can fluoresce if excited with right wavelength
GFP gene can be used for
gene for GFP can be fused to the gene of the protein of interest
Those genes would get expresses as fluoresced molecules
Helps monitor movement/localization of the protein
Lots of GFP colours (not just green) and have different wavelengths
How can a protein have a tuneable fluorescence (being able to control the fluorescence)
GFP has a beta barrel structure with 11 b-sheets, and a alpha-helix with the covalently bonded chromophore
The tightly packed nature of the barrel excludes solvent molecules, so it protects the chromophore (that fluoresces) from quenching (protected from water)
How can electrophoresis be used for protein analysis
Can measure mass (estimate) by looking at subunits of proteins
- separation is done by electrophoresis
- electric field pulls proteins according to their charge
- gel matrix hinders mobility of proteins according to their size and shape (large proteins move slower)
- SDS is a detergent (micelles bind and unfold proteins, give proteins negative charge)
- native shape does not matter
- Rate of movement only depends on size
SDS-PAGE properties/functions + reducing agents
SDS is very negative
Denatures proteins
Reducing agents: b-mercaptoethanol or DTT breaks disulfide bonds
What happens when SDS and a reducing agent are added to a protein sample?
When SDS and a reducing agent is added to a protein sample, the proteins are separated only based off the size of individual polypeptide chains