Carbohydrate Metabolism - Biochem Genetics
1/34
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
35 Terms
Carbohydrates
Carbohydrate: (CH20)n → saccharide monomers
Primary source for energy (ATP production)
Complex carbohydrates: longer saccharide chaines —> bread, legues, veggies
Simple carbohydrates: contain mono and disaccarhdires —>
Saccharide
Single “sugar” molecules
Ex: Glucose, Fructose, Galactose
Ring shaped: size of ring, orenation of the OH group
Disaccharides
Two linked sugar molecules
Ex sucrose(glucosle+furctose), Lactose (glucose +galactose), Maltose (Gluclose +glucose)
The common disaccharides (sucrose, lactose, maltose) are digested on the surface of the small insteintes—> enzymes excist: sucrase, Lactase, maltase
Break down into their mon saccharide comepents to allow for absortion through the GI wall and into the blood stream
“LActose intolerance”: loss of lactase activity in the small intestine; unable to be abasorded leading to bloating and discofmrt
Polysaccharides
Long chains of linked sugar molecules
Ex: glycogen (Animals), Amalose, Amolopectin, Cellleose (Plants)
Primary storage source for Gluclose in Animals and plants
Can also have structural functions (cellulose +chtitin)
Composed of diffrent monomeric units linked by different alpha+beta bonds
Starch
Main storage of glucose in plants: as either Amylose or amylopectin
Amylose is unbranched polysaccharide chain: linked only by alpha 1,4 linkages → long, uninterrupted chain of glucose (HELICAL SHAPE)
Amylopectin: have 1,4 alpha linkages chain with additional 1,6-alpha linkages of glucose → creating a branched molecule, higher storage capacity
Digestion starts with chewing, but needs
Salvitory gland → amylase
Pancreas → Amylopectin
Glycogen
Major storage form of glucose: highly branch a 1,4 and a 1,6 bonds
Body need a constant supply of blood glucose: taken in through diet but then stored:
Liver synthesizes and stores glycogen (glycogenesis): a repository for when glucose is low (between meals, sleep, fasting)
when blood glucose is low, liver glycogenolysis occurs
Glycogen → Glucose-6-Phostpaht (cannot leave liver)→ Glucose (CAN leave liver)
Glycogen storage= 8-10 hours of glucose → longer than that, switch to fat stores for days/weeks of energy (creates KETONE BODIES for energy)
Muscle has glycoen but no glu-6-phospahtase: → muslces can absord, store and use glyocen but cannot RELEASE IT BACK
Brain has no glycogen stores!! needs liver to provide a steady stream of gluclose
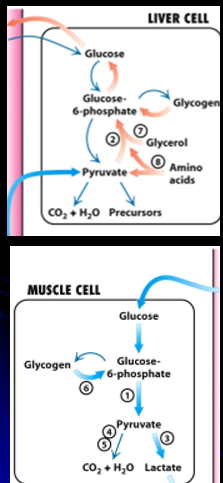
Gluconeogenesis
the process of synthesis of glucose within cells
this can use a number of different sources:
Amino Acids (call Glucogenic Amino Acids) : must be converted into substrates that can be metabolized to create glucose
Glycerol: created form fatty acid metabolism
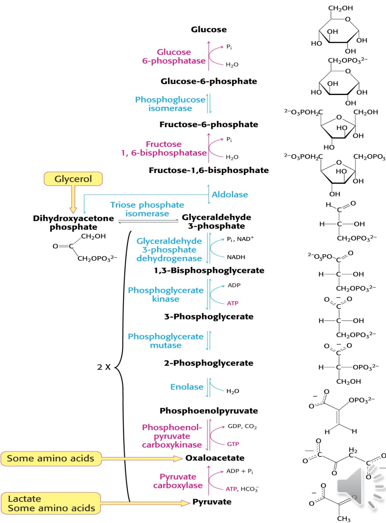
Glycolysis
process of metabolism glucose into energy intermediaries that can enter into Krebs cycle and oxidative phosphorylation for ATP production
Glu-6-phosphate → Pyruvate → Actyl CoA (which enter the krebs cycle)
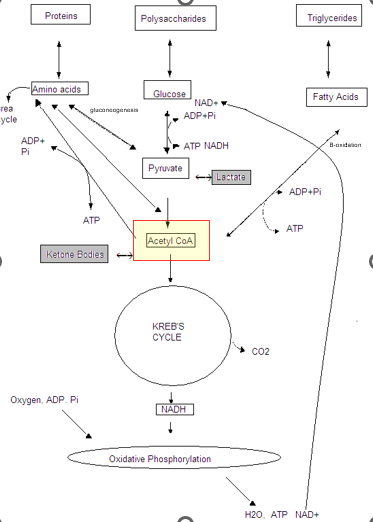
Ketones (Ketoacids)
Appear in hypoglycemic states, where the body shifts to Fatty Acid Beta-oxidations pathway to produce ACETYL -CoA
Ketone bodies can freely cross cell membranes —> CAN BE RELEASED INTO THE BLOOD STREAM as an alternative energy source for brain and muscle
Examples:
Acetoacetyl CoA
Acetone
B-OH butyrate
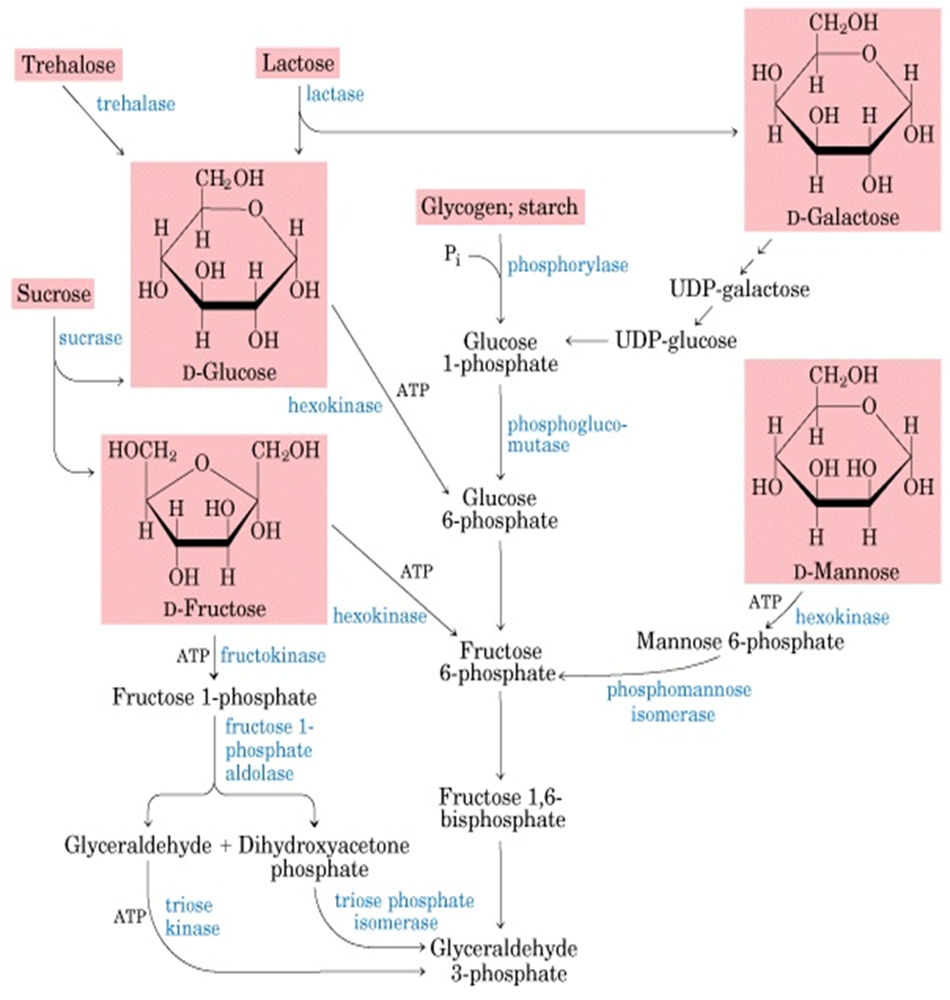
Galactose Metabolism
Lactose (present in breast milk) → Galactose + Glucose [Small Intestine]
Galactose → Galactose-1-phospahte via GALK → Gluclose-1-phosphae via GALT + co-factor UDP-glucose
UDP-glucose is converted to UDP-galactose and must be reconverted back to UDP glucose
Blocks in this system =
build up the different pathway products
Excess accumulation of galactose = galactic acid + galactitol
3 Galactosemia
3 major IEM
Galactokinase deficiency (GALK)
GALT deficiency → CLASSIC Galactosemic
GALE Deficiency
Autosomal Recessive
Galactokinase deficiency: untreated leads to cataracts → Galatonic acid + galactical build up as deposits in the eye
Gale Deficiency: Mild + Severe (rarer, galactosemia-like) → high gal-1-phosphate and galactose + UDP galactose
ALL treted with DIetary Galactose restiction: but glactose also naturally produces galactose on its own so not fully sufficeint
Classic Galactosemia
GALT Deficiency
Neonatal onset: when baby is introduced to breast milk: Glaonic acid/Galctocla buildup
Lethargy, poor feeding, vomiting, diaheria, death (LPVDD)
Liver Failure: jaundice, bleeding, liver swelling (hepatomegly from gal-1-p buildup)
Renal dysfunction
Cataracts (galactitol buildup)
E Coli (gram negative) sepsis": flourishes in environment w high galactose
Labs
Elevated liver enzymes and bilirubin
Coagualopathy (liver dysfunction)
Positive reducing substance in urine
Urine Galactose
All monosaccharides are reducing sugars → Will reduce inorganic ions such as copper via Fehling’s reagent
Urine testing will detect presence of reducing sugars in urine (normally DOES NOT HAPPEN)
thin layer chromatography (TLC) can distinguish between different sugars that are present
Blood Glucose and GALT
Newborn screening relies on the blood spot measurement of Galactose and Gal-1-P levels
CAN DETECT ALL 3 Galactosemias
But false negative: infants on lactose-free formulas → wont accumulate enough
Alternative strategy: Detection of GALT activity
Prost-transfusion false negative
Detects Galatsemia classic, but misses the other two
Galt activity vs measuring Galactose and Gal-1-P levels
Galactosemia Treatment
Lifelong Galactose free diet: No breast milk, No Soy
Outcome with Treatment
Life threating disease resolves quickly
Long term issues: we will endogenously produce galactose even if it is fully removed from the diet
reduced IQ + growth,
ataxia,
ovarian dysfunction
GALT Mutation
Q188R in Caucasian 70% of classical cases
S135L in 65% of African AMerican “Clinical variant”
can have better outcomes: better ovarian function
harder to detect: better GALT function
N314D (Durate variants) → 50% GALT activity reduction
two Duartes mutations = GALT 50% activity, are just ‘carriers’
Fructose Metabolism
Fructose mainly produced via the breakdown of Sucrose:
Fructose Disorder
3 Main Disorders of Fructose Metabolism
Fructokinase Deficiency (Fructosurea)
Hereditary Fructose Intolerance (Fructosemia)
Fructose 1,6-Biphosphatase Defcieny
All autosomal recessive
Fructokinase Deficiency (Fructosurea)
Benign condition
Usually detected indinally
Urine reducing substance: Fructose
No treatment necessary
Hereditary Fructose intolerance
Etiology: Gene for Fructose-1-P Aldolase B (ALDOB gene) → a deficiency
Symptoms: only once diet includes fructose → sigfnicant reactions
vomiting, lethargy, irritability, seizures
Postprandial hypoglycemia, liver/kidney Dz
Liver failure, acidosis, growth failure, death
Diagnosis
Suspect with urine reducing substance for fructose:
Fructose + fructose-1-phosphae (toxic for body + decrease glycolysis)
confirm with enzyme activity + mutation analysis
Treatment
Eliminating fructose prevents and reverses further symptoms
So patients will remain resistant despite elimination of fructose
Fructose 1,6-Biphosphatase
Symptoms: sudden, early life-threatening episodes of:
Fasting hypoglycemia with lactic acidosis (***so even when Fructose has been eliminated***) → inability to shunt over Gly-3-P into the glycogensis pathway
hyperventilation, vomiting, lethargy
may be lethal
Fructose 1,6-Bisphosphatase → Glyceryalhyd-3-Phosphate (usable for glycognesisa) via 1,6-Bisphosphatase activity
Diagnosis
NO URINE FRUCTOSE
Check enzyme on liver biopsy
Urine glycerol-3-phosphateleves
Molecular testing
Treatment
Acute: correct hypoglycemia and acidosis, IV
Chronic: Avoid fasting with frequent glucose feeding, limit fructose
Good outcomes once treatment initiated
Glycogen Storage Disorders
When glycogen cannot be broken down, it accumulates in the cells they are stored in:
Liver involvement: causing hypoglycemia (inability to liberate glucose from glycogen) and swelling of the liver
Muscle involvement: causing muscle breakdown and weakness
Almost all are Recessive, but one if X-linked
Type 1 Von Gerike Diease
Type 2 Pompe Diease
Type 5 McArdle Disease
Von Gierke Disease (GSD1) Metaboism
Gluclose-6-phosphatase deficiency in Liver, Kidney and intestinal mucosa
Type1a: the glucose-6-phsopatase enzyme is defective
Type 1b: membrane transport protein for gluclose-6-phosphate
When fasting, Body needs Glu-6-phosphate to be converted to glucose
GSD1 Both type susceptible to Hypoglycemia when fasting
Von Gierke (GSD1) Symptoms
Usually not present early on: WELL FED = protection from fasting and therefore hypoglycemia
Sometimes hypoglycemia and lactic acidosis
Usually present at 3-4 months of age
Hepatomegaly
Hypoglycemia
Hyperuricemnia
Hyperlipidemia (fats are broken down more and end up in the blood)
Lactic acidosis
“Doll-Like” faces with fat checks, relatively thin extrmeties, short statures and protrubent—> weird fat deposits
Type Ib patients also have neutropenia
Von Gierke (GSD1) Diagnosis
Suspicion: usually infants more than new-born→ NOT ON NEWBORN SCREENING
Hypoglycemia with minimal fasting
Lactic acidosis, hyperuricemia, hyperlipidemia
Diagnosis
Enzyme analysis: requires live biopsy
Mutation Analysis
GSD1a: Glu-6-Phosphatase (G6PC) gene (94%)
Ashkenazi gene
GSD1b: Glu-6-Phosphate translocase (SLC37A4) gene (95%)
PRESENTS SIMILARLY TO GSDII: but diffrent genes, more milder and invovles MUSCLEs (myopathy)
Von Gierke (GSD1) Treatment
Avoid fasting/maintain blood glucose
May need continuous nasogastric glucose
Uncooked cornstarch: slow-release of glucose → when their salivatory gland mature enough to release amylase (complex carbohydrate breakdown)
Restrict Fructose and galactose (cannot be converted to free glucose)
Dietary supplments
Allopurinol for uric acid
Statins for cholesterol
Watch for hepatic adenomas
G-CSF for neutropenic immunodeficiency
Pompes Disease (GSDII) Metabolism
Mechanism:
Lysosomal Acid-alpha-Glucosidae (acid maltase) (GAA) Deficiency —> Lysosomal Glycogen Storage
Forms
infantile: more severe → cardiomegaly, liver swelling
Juvenile + Adult onset → less severe
Pompe’s Deasiae (GSDII)
Organs affected by glycogen accumulation
Cardiac, skeletal and smooth muscle (primarily)
Muscle and Organ enlargement (due to Glycogen over-storage)
Tongue (macroglossia)
Heart (cardio Meagley)
Liver
Spleen
Progressive Muscle Breakdown and Weakness
Elevated CPK
Hypotonia
hypertrophic cardiomyopathy
Cardio-respiratory failure
Pompe’s Disease (GSDIII) Diagnosis
Newborn screening: <10 days in IOPD (enzyme, DNA, CRIM, echo, CK)
Muscle biopsy with glycogen staining, enzyme, and DNA
Pseudo-deficiency: low enzyme but asymptomatic → check DNA
Pompe’s disease (GSDIII) Treatment
Lysomal enzyme replacement therapy:
Outcomes:
Without treatment: Death within on year
With treatment: improvement of symptoms and delayed onset (in infantile)
Does not stop all symptoms
McArdle Disease (GSD V)
No infantile onset + asymptomatic throughout infancy and childhood NT PROGRESSIVE, but EPIDOSIC → after prolonged periods of muscle excertion (pains + weakness after exercising)
Mutation in the muscle specific enzyme: Muscle Myophosphorylase (PYGM)
→ Decreased glycogenolysis by decreasing the cleavage of branches from the branched glycogen chain
No hypoglycemia: Not important in the liver for the liberation of glucose from the branched glycogen molecule (other enzyme in the liver able to do that)
But not the CASE IN MUSCLES
McArdle Disease (GSD V) Symptoms
Initial Presentation
recurrent excersice induced muscle cramps and pain releaved by rest
Recurrent epsidoic myoglobinurea → can lead to Renal Failure in severe cases
Often lifelong history of poor escerise capacty
Chronic proximal muscle weakness
McArdle Disease (GSD V) Diagnosis
Screening
Elevated Creatine Kinase (CK) levels after exercise
Urine myoglobin levels
Diagnosis
Enzyme analysis: Myophopshalyase activity on muscle biopsy
Molecular analysis: PYGM gene sequencing + del/dup analysis
Similar to GSD VII (Tarui’s Diease)
But diffrent gen and intial slow growth and anemia not seen in GSD V
McArdle Disease (GSD V) Treatment
Sub-maximal aerobic excercise: avoidn maximal aerobi or isometric excerise
Avoid medicaitons that promote myopathy: statins, certina anesthetic
Diet and supplments
Creatine monohydrte supplemnation
Carbohydrate rich diet
Pre-exercise simple sugars
Classic GLUT1 Defecieny
Glucose Transport protein deficiency: diffuclty transporting gluclose acrossthe blood-brain barrier
Normal levels of glucose in the blood stream but low in CS fluid→ energy deficiency in the brain
AUTOSOMAL DOMINANT SLC2A1 gene mutation
Symptom
Infaitlne seziures
delveomntl delap,microcehpyl
complex movement disorder
Treatment results in significant improvment:
Ketogenic diet: allow for alternaive use for engery than GLUCLOSE
L-Carnitine
Avoid glucose intake