Exam 1 review Genomics
1/95
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
96 Terms
De Novo assembly
Building the genome from scratch only using the reads you sequenced
No reference genome
Needs high coverage (sequence more)
For a de novo assembly, using Illumina, how much coverage would you need?
~80x
For a de novo assembly, using pacbio how much coverage would you need?
~20-30x
Reference- guided assmebly
You already have a closely related genome to help guide the assembly.
The existing genome acts like a map to help put the puzzle pieces together faster and more accurately.
Needs less coverage because you have help:
~30-50x for Illumina
When would you use reference-guided assembly instead of de novo?
When you have a closely related genome to use as a guide — it’s faster and requires less data.
Why is high coverage needed for de novo assembly?
To make sure you have enough overlapping reads to piece the genome together accurately.
Which sequencing technology is better for assembling complex genomes from scratch?
PacBio or Oxford Nanopore (long reads)
If you wanted to analyze gene expression across samples, which sequencing method would you use?
Illumina (short-read) is most commonly used
Which platform is best for identifying small mutations like SNPs?
Illumina for SNPs
Why are long reads useful for de novo assembly?
Because they can span large repetitive regions, making assembly easier and more accurate.
Can you sequence a full human genome at 30X on one Illumina flow cell?
No — one run gives 0.625 of a full genome
What would happen if you only had 10X coverage?
Your data might be less accurate — harder to detect mutations or assemble the genome.
Global Alignment
What it does: Aligns the entire length of two sequences from beginning to end.
Best when:
Sequences are similar in length
Sequences are closely related
Local Alignment
What it does: Finds the most similar region between sequences. Doesn’t try to align everything — just the best matching parts.
Best when:
Sequences are not the same length
Only parts of them are similar
Good for finding conserved motifs/domains
Which alignment type is better for comparing short reads to a reference genome?
Global allighment
If you were comparing a new protein to a huge database of unrelated sequences, which method should you use?
Local alignment — to find the most similar regions
Why local alignment is usually more useful:
Most real-world biological sequences are not 100% the same
Only parts might be conserved (similar across species)
Mutations, insertions, deletions make full alignments messy
Local alignment focuses on the good parts only
Sequences are distantly related
You're looking for a motif or conserved domain inside a larger sequence
You’re aligning a short read or fragment to a long reference genome
Can global alignment still produce full-length results if the sequences are highly similar?
Yes! If the sequences are similar and of equal length, global alignment can work well
What is something unique to Global Alignment when trying to match sequences?
Global alignment tries to match every position, even if it means adding gaps to make things fit.
Why is global alignment better for short reads?
Because short reads (like from Illumina) are already:
Very short (usually 100–300 base pairs)
Designed to match a reference exactly or almost exactly
From a known location in the genome you're sequencing
What tool uses local alignment to compare sequences in a database?
BLAST
What does a progressive Multiple Sequence Alignment do?
It aligns sequences one at a time, adding each new sequence to the existing alignment
Why use multiple sequence alignment?
To identify conserved regions and study evolutionary or functional relationships
Like building phylogenetic trees
What’s one challenge with MSA?
Errors made early on can affect the final alignment, especially if sequences are very different

What is a consensus sequence?
A sequence that shows the most common base/amino acid at each position from a multiple alignment
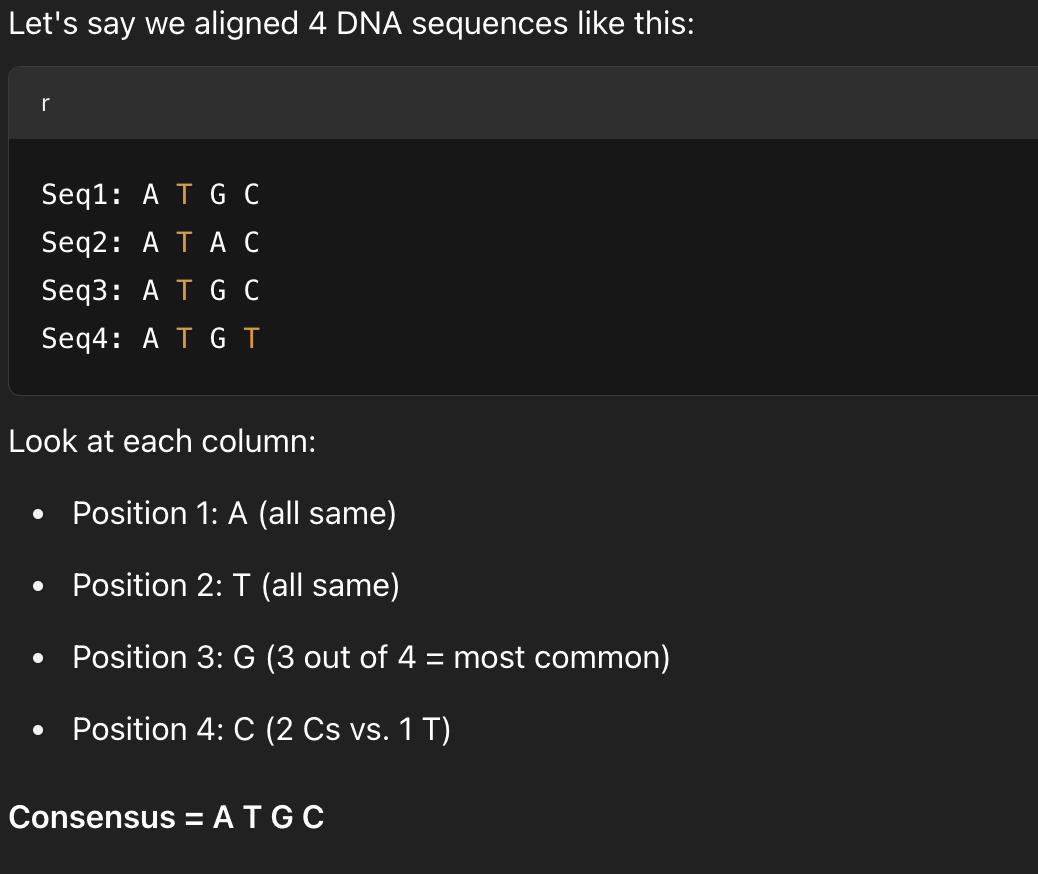
What does a consensus sequence help identify?
Conserved and functionally important regions shared across multiple sequences
It helps identify regions that are evolutionarily conserved or functionally critical
What is the molecular clock used for?
What kind of mutation does the molecular clock rely on?
To estimate how long ago two species or genes diverged from a common ancestor
Neutral mutations that accumulate at a constant rate
How is the rate of evolution calculated?
By dividing the number of sequence changes by the time since divergence
BLOSUM (BLOcks SUbstitution Matrix)
Based on observed substitutions in blocks of real protein sequences
Used for local alignment (e.g., in BLAST)
BLOSUM number = % similarity cutoff
BLOSUM80 = for closely related proteins (strict)
BLOSUM62 = standard default
BLOSUM45 = for distantly related proteins (more tolerant)
Lower number = more tolerant of change
PAM (Point Accepted Mutation)
Based on evolutionary models — how proteins evolve over time
Used more for global alignments
PAM number = evolutionary time
PAM30 = short time, very similar sequences
PAM250 = long time, lots of changes allowed
Higher number = more time has passed = more differences allowed
What does BLOSUM62 mean?
It’s a scoring matrix based on proteins with up to 62% similarity — good for moderately related sequences
Which matrix would you use to compare distantly related proteins?
BLOSUM45 or PAM250 — they tolerate more differences
Substitution rate
how often one amino acid replaces another
Ungapped alignment
sequence alignment without gaps (used in BLOSUM building)
Which matrix would you use to align very similar protein sequences?
BLOSUM80 or PAM30
FASTA Format
Used for: storing sequences without quality scores
Mostly used for:
Assembled genomes
Contigs
Protein sequences
FASTQ Format
Used for: raw sequencing reads with quality scores
Used during preprocessing, assembly, and alignment because quality matters at that stage
What is the E-value?
It tells you how likely it is to get that match just by chance.
A low E-value = very unlikely the match happened randomly → good hit!
A high E-value = could just be a fluke → probably not meaningful
What is the Bit Score?
t’s a number based on the raw alignment score (matches, mismatches, gaps)
But it also accounts for the scoring system and statistical background
Because it’s normalized, you can compare bit scores across different searches, even with different databases or matrices
Why use both E-value and Bit Score?
E-value tells you how likely the match is real
Bit score tells you how strong the match is
Was the genome assembled into one piece? for yersinia pestis?
No.
The reference-guided assembly made 130,556 contigs (pieces of genome)
The N50 was only 288 bp → this means most contigs were very small
This tells us ancient DNA was highly degraded, so they couldn’t stitch it together cleanly
Was increased virulence due to mutations?
No unique mutations were found in the medieval DNA that aren’t also found in modern strains
So, the extreme death toll was probably not due to the bacteria being more deadly genetically
What might have caused the high death toll if not bacterial mutations?
Environmental conditions, vector behavior, and host vulnerability
What does a low N50 tell us about a genome assembly?
It means the assembly is fragmented — mostly short contigs
Why don’t scaffolds always cover the whole genome perfectly?
Gaps can result from low coverage, repetitive regions, or sequencing errors
Why use OLC?
Overlap – finding matching regions between reads
Layout – ordering and connecting those reads
Consensus – choosing the final base sequence from the overlaps
Good for long reads like pacbio or nanopore, slow for short reads tho
Why are de Bruijn graphs efficient for short reads?
Because they avoid all-vs-all read comparison and just work with k-mer overlaps
What’s a drawback of using de Bruijn graphs?
They are sensitive to sequencing errors and lose long-range information
Which method better resolves long repetitive regions? olc/dbg
OLC — because long reads can span the repeats
What type of assembly method do most modern genome projects use? (olc/dbg)
Hybrid assembly — combining short and long read strategies
It combines the accuracy of short reads with the long-range context of long reads for better genome coverage
N50 calculation and logic
Total genome = 100 million bp
You add contigs from longest to shortest:
30 Mb, 15 Mb, 10 Mb, 7 Mb, 5 Mb…
You stop once you’ve added 50 million bp
The shortest contig used to reach that halfway mark is your N50
What is the difference between N50 and NG50?
N50 is based on the assembly size; NG50 is based on the known/reference genome size
If your N50 is very low, what does that mean about your assembly?
It’s fragmented — made up of mostly short contigs
What is an inversion in genome assembly?
A region that is flipped in direction compared to the reference
What does a high mismatch rate indicate?
Many single-base differences from the reference — possibly sequencing or assembly errors
What is a translocation error in genome assembly?
A sequence is assembled into the wrong location — possibly the wrong chromosome
what do you want to do before processing reads in an assemble like SPAdes (DbG assembler)?
Pre-process reads by trimming low-quality sequences
remove adapters and contaminants
remove human contaminants
normalize or subsample to even out read coverage (if you have too many reads)
error correction (fix base mistakes)
Why might long k-mers fail in some assemblies?
If coverage is too low, longer k-mers can’t find overlaps and assemblies become fragmented
What’s an ab initio method?
A computational method that predicts genes based on sequence patterns and signals
What do homology-based methods rely on?
Sequence similarity to known genes in other species
Why is RNA-seq useful for gene finding?
It provides experimental evidence of which DNA regions are being actively transcribed
Which method is best when no related genome is available when it comes to gene finding?
Ab initio methods
Gene annotation
Once you’ve assembled a genome and maybe identified genes, you still need to label what those regions do. That’s annotation — and it can be done in two ways:
manual or computational
Manual Annotation (by experts):
one by trained scientists, often using genome browsers (like UCSC or Ensembl)
They look at:
Sequence context
Homologs
Expression data
Functional evidence
Pros and cons of manual gene annotation
✅ Pros:
Accurate (humans can catch weird or subtle things computers miss)
❌ Cons:
Very slow
Labor-intensive
Results can be inconsistent between people
Computational Annotation (fully automated):
Uses algorithms to assign gene functions, locations, structures, etc.
May use ab initio models, homology, or RNA-seq data
Computational Annotation pros and cons
✅ Pros:
Fast and consistent
Great for annotating many genomes at once
❌ Cons:
Can be wrong or imprecise (e.g., missing start codons, splitting/merging genes incorrectly)
What’s the recommended strategy for genome annotation?
Combine computational methods with manual curation
Why is ab initio gene prediction easier in prokaryotes?
Because their genes don’t have introns and are often long, continuous ORFs
How do ab initio methods work in eukaryotes despite introns?
They use models based on sequence patterns like nucleotide composition and codon usage
Which gene elements are hard to predict computationally?
Promoters and UTRs
Which approach can provide a full TU?
Evidence-based gene finding (e.g., RNA-seq), but not promoters
Why is promoter prediction unreliable?
t requires experimental data to detect transcription start sites accurately
BLASTP = Protein BLAST
It compares your unknown protein to known proteins in a database
If your sequence looks a lot like a protein with a known function → you can guess its function
Reference Genome Approach: Annotation
Use when you have a closely related organism with a well-annotated genome
You compare your protein to the proteins in that organism’s database
If it's very similar, you can even use DNA-level comparisons
Universal Protein Databases
Use if no good reference genome exists
These databases have highly annotated proteins from many species
Popular databases:
✅ SwissProt – curated, very high quality
✅ UniProt – big database, includes SwissProt + unreviewed entries
✅ NCBI nr (non-redundant) – massive, less curated, but very inclusive
What is functional gene annotation?
Assigning a likely biological function to a predicted gene or protein
Why are domains important?
They tell you what a part of the protein does, even if the whole protein is new
They’re more evolutionarily stable than full sequences
Think of them like Lego blocks used in different proteins
What conclusion can you make if a protein has a receptor domain and a kinase domain
It may function as a receptor kinase, signaling from the outside to the inside of the cell
What is a motif in protein annotation?
A short, highly conserved sequence important for function, often part of the catalytic center
Ontology (in bioinformatics)
a fancy way to describe what parts exist in a biological system and how they’re related, in a way computers can understand.
Think of it like a biological family tree or mind map, but for:
Genes
Proteins
Cell parts
Functions
Who maintains the most widely used ontology in biology?
The Gene Ontology Consortium
What is a DAG?
AG = Directed Acyclic Graph
↳ It’s like a hierarchy, but better:
- Arrows show direction (like from general to specific).
- It doesn’t loop back (acyclic).
- A term can have multiple parents (not just one!).
What does “part_of” mean?
It means one term is a component of another, but the bigger part doesn't always require the smaller one to exist.
Robust (Strong Experimental Evidence):
These evidence codes come from direct biological experiments — trusted!
Less Robust (Computational or Indirect):
These evidence codes may be useful, but they rely more on inference or author notes — less reliable.
Gene-to-GO mappings aren't unique why?
→ One gene can be linked to many GO terms (within different or even the same ontology categories: molecular function, cellular component, or biological process).
What’s a downside of in silico GO annotations?
They are often lower quality and less reliable than manually curated annotations.
What does RNA-Seq measure?
It measures the level of gene expression across the genome.
What Must Be Normalized? to compare RNA-seq expression?
To compare expression fairly, we must adjust for:
Sequencing Depth – Did one sample just get sequenced more?
Transcript Length – Longer genes get more hits just by size.
RNA Composition – Highly expressed genes can dominate the total reads.
Why can’t you directly compare raw read counts between genes? (RNA-seq)
Because differences might be due to gene length, sequencing depth, or RNA composition, not actual gene expression.
What does TPM stand for and why is it preferred?
TPM = Transcripts Per Million. It's preferred because it allows for fair comparisons across samples and the total expression is normalized to a consistent scale.
When would you use RPKM instead of TPM?
Historically, RPKM was used for single-end sequencing, but TPM is now favored even in those cases due to better comparability.
he Problem with RPKM:
normalizes for:
gene length ✅
sequencing depth ✅
BUT…
It does NOT ensure that the total expression across all genes in a sample is the same.
So, if two samples have different total transcript amounts (like in a disease vs healthy state), RPKM values can be distorted and not comparable across samples.
methods of ways to normalize
Goal | Best Method |
|---|---|
Comparing expression levels within a single sample | TPM |
Comparing expression levels between samples or conditions | DESeq2 (or EdgeR/TMM) |
Visualizing general abundance, not for stats | TPM |
Accurate DE analysis (statistical tests) | DESeq2 (requires raw counts + proper transformation) |
Use CPM when:
You're comparing gene expression between replicates of the same group (e.g., how consistent is Gene X in 3 untreated samples?).
You want a quick visualization or filtering lowly expressed genes before DE analysis.