Transformation & Conjugation
1/64
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
65 Terms
Discuss the source, natural function, structure and uses of restriction enzymes.
Explain how recombinant DNA is made.
Describe the effect of the following on electrophoresis: voltage gradient, current, gel concentration, charge density, pH.
Enunciate the major features of the different types of cloning vectors and their use.
Describe how to make a genomic and a DNA library of a given species.
Mention some clinical uses of recombinant DNA.
all the learning objectives have been done individually.
need to go to textbook for clinical correlation.
Discuss the source, natural function, structure and uses of restriction enzymes.
Restriction enzymes, also known as restriction endonucleases, are one of the most important tools in molecular biology. Here is a detailed discussion of their source, natural function, structure, and uses.
1. Source
Restriction enzymes are naturally found in bacteria (and archaea).
Bacterial Origin: Virtually every species of bacteria produces one or more specific restriction enzymes. For example, the well-known enzyme EcoRI is isolated from the R strain of the bacterium E*scherichia coli.
2. Natural Function: The Bacterial Immune System
In their natural bacterial context, restriction enzymes serve as a primitive immune system that protects the bacterium from infection by foreign DNA, most commonly bacteriophages (viruses that infect bacteria).
This system is known as the Restriction-Modification System and consists of two components:
The Restriction Enzyme (The "Scissors"): This enzyme cuts DNA at specific, short sequences.
The Methyltransferase Enzyme (The "Marker"): This enzyme modifies the same specific sequences in the bacterium's own DNA by adding a methyl group (e.g., to an adenine or cytosine base). This methylation marks the host DNA as "self."
How it works:
When a bacteriophage injects its DNA into a bacterial cell, the DNA is unmodified ("non-self").
The bacterial restriction enzyme scans the incoming viral DNA for its specific recognition sequence and, upon finding it, cuts the DNA into fragments, destroying it and aborting the infection.
The bacterium's own DNA is protected from being cut because the same sequences are methylated by the methyltransferase.
3. Structure
Restriction enzymes are proteins that recognize and bind to specific DNA sequences. Their structure is key to their function.
General Form: They are typically dimers (often homodimers), meaning they are composed of two identical protein subunits. This structure is perfectly suited for recognizing palindromic sequences.
Recognition Site: The DNA sequence they recognize is usually palindromic, meaning the sequence of nucleotides is the same on both strands when read in the 5' to 3' direction.
Example: The recognition site for EcoRI is 5'-GAATTC-3'. Read the complementary strand, also 5' to 3': 5'-GAATTC-3'. They are identical.
Active Site: The enzyme's active site, where the cutting occurs, is positioned to catalyze the hydrolysis of the phosphodiester bond in the DNA backbone.
There are three main types (I, II, and III), but Type II is the most important for molecular biology:
Type II Restriction Enzymes:
Function: These enzymes are the workhorses in the lab. They are simple and predictable: they cut at defined locations within or very close to their recognition sequence.
Cutting Patterns: They can produce two types of ends:
Sticky Ends (Staggered Cuts): The enzyme cuts the two DNA strands at different points, creating short, single-stranded overhangs.
Example: EcoRI cuts between G and A, producing 5' overhangs.
5'...G A-A-T-T-C...3'→5'...G&A-A-T-T-C...3'3'...C-T-T-A-A G...5'→3'...C-T-T-A-A&G...5'
Blunt Ends (Straight Cuts): The enzyme cuts both DNA strands at the same point, creating no overhang.
Example: EcoRV cuts in the middle, producing blunt ends.
5'...G-A-T-A-T-C...3'→5'...G-A-T&A-T-C...3'3'...C-T-A-T-A-G...5'→3'...C-T-A&T-A-G...5'
4. Uses: The Foundation of Recombinant DNA Technology
The discovery of restriction enzymes made genetic engineering possible. Their primary use is to cut DNA molecules in a precise and predictable manner.
A. DNA Cloning
Process: A restriction enzyme is used to cut both the vector (e.g., a plasmid) and the DNA fragment of interest (e.g., a gene).
Ligation: If the same enzyme (or one that produces compatible ends) is used, the complementary "sticky ends" of the vector and the insert will anneal. Another enzyme, DNA ligase, is then used to permanently seal the fragments together, creating a recombinant DNA molecule.
Application: This recombinant plasmid can be inserted into bacteria, which then multiply, producing millions of copies (clones) of the gene for study or for producing a protein (like insulin).
B. Gene Analysis and Mapping
Restriction Fragment Length Polymorphism (RFLP): Slight variations (polymorphisms) in DNA sequences between individuals can create or destroy restriction enzyme recognition sites. When cut with a specific enzyme, these individuals will produce DNA fragments of different lengths. RFLP was a foundational technique for genetic fingerprinting, disease gene mapping, and paternity testing.
Restriction Mapping: By using a series of different restriction enzymes (alone and in combination) on a DNA molecule, researchers can deduce the order and relative distances between the cut sites, creating a "map" of the DNA.
C. Molecular Diagnostics
Principle: The presence or absence of a specific restriction site can be used to diagnose genetic diseases.
Example: Sickle Cell Anemia is caused by a single base-pair mutation (A to T) in the beta-globin gene. This mutation also destroys a recognition site for the restriction enzyme MstII. By performing a restriction digest on a patient's DNA sample, the disease can be diagnosed based on the size of the resulting fragments.
D. DNA Sequencing and Southern Blotting
Library Construction: Restriction enzymes are used to fragment genomic DNA into manageable pieces for sequencing (e.g., in the original Human Genome Project).
Southern Blotting: This technique involves cutting DNA with restriction enzymes, separating the fragments by gel electrophoresis, and then using a labeled probe to identify a specific fragment, allowing for the detection of a specific gene sequence.
Summary Table
Aspect | Description |
|---|---|
Source | Bacteria and archaea. |
Natural Function | Part of the Restriction-Modification system; defends against bacteriophage infection by cutting non-methylated foreign DNA. |
Structure | Typically dimeric proteins that recognize short, palindromic DNA sequences (4-8 base pairs). |
Key Types | Type II is most useful; cuts at a defined site within its recognition sequence. |
Primary Use | To cut DNA at specific sequences to create recombinant DNA molecules for cloning. |
Other Uses | Genetic fingerprinting (RFLP), gene mapping, molecular diagnostics, and DNA sequencing. |
In essence, restriction enzymes are the "molecular scissors" that allowed scientists to move from simply studying DNA to actively manipulating it, launching the era of biotechnology and modern molecular medicine.
Restriction enzymes, also known as restriction endonucleases, are one of the most important tools in molecular biology. Here is a detailed discussion of their source, natural function, structure, and uses.
1. Source
Restriction enzymes are naturally found in bacteria (and archaea).
Discovery: They were first discovered and characterized in the 1960s and 1970s, with Werner Arber, Hamilton Smith, and Daniel Nathans sharing the 1978 Nobel Prize in Physiology or Medicine for their work.
Bacterial Origin: Virtually every species of bacteria produces one or more specific restriction enzymes. For example, the well-known enzyme EcoRI is isolated from the R strain of the bacterium E*scherichia coli.
2. Natural Function of restrictive enzymes: The Bacterial Immune System
restriction enzymes serves as a primitive immune system that protects against bacteriophages (viruses that infect bacteria)
This primitive immune system is known as the Restriction-Modification System and consists of two components:
The Restriction Enzyme (The "Scissors"): This enzyme cuts DNA at specific, short sequences, destroying bacteriophage dna.
The Methyl-transferase Enzyme (The "Marker"): This enzyme modifies the same specific sequences in the bacterium's own DNA by adding a methyl group (e.g., to an adenine or cytosine base). This methylation marks the host DNA as "self."
How it works:
1.When a bacteriophage injects its DNA into a bacterial cell, the DNA is unmodified ("non-self").
2. The bacterial restriction enzyme scans the incoming viral DNA for its specific recognition sequence and, upon finding it, cuts the DNA into fragments, destroying it and aborting the infection.
The bacterium's own DNA is protected from being cut because the same sequences are methylated by the methyltransferase
3. Structure of restriction enzymes
Restriction enzymes are proteins that recognize and bind to specific DNA sequences. Their structure is key to their function.
General Form: homodimers
The structure of restriction enzymes (homodimers) are perfectly suited for recognizing palindromic sequences.
Recognition Site: The DNA sequence they recognize is usually palindromic, meaning the sequence of nucleotides is the same on both strands when read in the 5' to 3' direction.
Example: The recognition site for EcoRI is 5'-GAATTC-3'. Read the complementary strand, also 5' to 3': 5'-GAATTC-3'. They are identical.
Active Site: The enzyme's active site, where the cutting occurs, is positioned to catalyze the hydrolysis of the phosphodiester bond in the DNA backbone.
There are three main types (I, II, and III) of restriction enzymes, but Type II is the most important for molecular biology:
Type II Restriction Enzymes:
Function: These enzymes are the workhorses in the lab. They are simple and predictable: they cut at defined locations within or very close to their recognition sequence.
cutting patterns (types of ends): restriction enzymes can produce two types of ends:
Sticky Ends (Staggered Cuts): The enzyme cuts the two DNA strands at different points, creating short, single-stranded overhangs.
Example: EcoRI cuts between G and A, producing 5' overhangs.
5'...G A-A-T-T-C...3'→5'...G&A-A-T-T-C...3'3'...C-T-T-A-A G...5'→3'...C-T-T-A-A&G...5'Blunt Ends (Straight Cuts): The enzyme cuts both DNA strands at the same point, creating no overhang.
Example: EcoRV cuts in the middle, producing blunt ends.
5'...G-A-T-A-T-C...3'→5'...G-A-T&A-T-C...3'3'...C-T-A-T-A-G...5'→3'...C-T-A&T-A-G...5'
The Foundation of Recombinant DNA Technology
DNA cloning
Gene analysis
Molecular Diagnostics
DNA sequencing
Uses of restriction enzymes: The Foundation of Recombinant DNA Technology
The discovery of restriction enzymes made genetic engineering possible. The primary use of restriction enzymes is to cut DNA molecules in a precise and predictable manner.
A. DNA Cloning
Process: A restriction enzyme is used to cut both the vector (e.g., a plasmid) and the DNA fragment of interest (e.g., a gene).
Ligation: If the same enzyme (or one that produces compatible ends) is used, the complementary "sticky ends" of the vector and the insert will anneal. Another enzyme, DNA ligase, is then used to permanently seal the fragments together, creating a recombinant DNA molecule.
Application: This recombinant plasmid can be inserted into bacteria, which then multiply, producing millions of copies (clones) of the gene for study or for producing a protein (like insulin).
B. Gene Analysis and Mapping
Restriction Fragment Length Polymorphism (RFLP): Slight variations (polymorphisms) in DNA sequences between individuals can create or destroy restriction enzyme recognition sites. When cut with a specific enzyme, these individuals will produce DNA fragments of different lengths. RFLP was a foundational technique for genetic fingerprinting, disease gene mapping, and paternity testing.
Restriction Mapping: By using a series of different restriction enzymes (alone and in combination) on a DNA molecule, researchers can deduce the order and relative distances between the cut sites, creating a "map" of the DNA.
C. Molecular Diagnostics
Principle: The presence or absence of a specific restriction site can be used to diagnose genetic diseases.
Example: Sickle Cell Anemia is caused by a single base-pair mutation (A to T) in the beta-globin gene. This mutation also destroys a recognition site for the restriction enzyme MstII. By performing a restriction digest on a patient's DNA sample, the disease can be diagnosed based on the size of the resulting fragments.
D. DNA Sequencing and Southern Blotting
Library Construction: Restriction enzymes are used to fragment genomic DNA into manageable pieces for sequencing (e.g., in the original Human Genome Project).
Southern Blotting: This technique involves cutting DNA with restriction enzymes, separating the fragments by gel electrophoresis, and then using a labeled probe to identify a specific fragment, allowing for the detection of a specific gene sequence.
Explain how recombinant DNA is made.
Creating recombinant DNA is a foundational technique in biotechnology, often called gene cloning. The process involves combining DNA molecules from two different sources to create a new, hybrid DNA molecule.
The core idea is to use two key tools:
Restriction Enzymes to cut DNA at specific sites.
DNA Ligase to paste DNA fragments together.
Here is a step-by-step explanation of how recombinant DNA is made, using the classic example of inserting a human gene into a bacterial plasmid to produce a protein like insulin.
Step-by-Step ProcessStep 1: Isolate the DNA of Interest and the Vector
DNA of Interest (Insert): This is the gene or DNA fragment you want to clone. For example, the human insulin gene. This can be obtained by:
Isolating mRNA from human pancreatic cells and using Reverse Transcriptase to create complementary DNA (cDNA).
Cutting it out of human genomic DNA using a restriction enzyme.
Synthesizing it artificially in a lab.
Vector: This is a DNA molecule that will carry the insert into a host cell (like a bacterium) and enable it to replicate. The most common vector is a bacterial plasmid—a small, circular, double-stranded DNA molecule that is separate from the bacterial chromosome. Plasmids often contain:
An Origin of Replication (ORI): So it can be copied inside the host cell.
A Selectable Marker: Often an antibiotic resistance gene (e.g., Ampicillin resistance), which allows us to later identify bacteria that have taken up the plasmid.
A Multiple Cloning Site (MCS): A short region containing recognition sites for many different restriction enzymes, making it easy to insert the DNA fragment.
Step 2: Cut Both DNA Molecules with the Same Restriction Enzyme
This is the most critical step for ensuring the pieces can be joined.
The same restriction enzyme (e.g., EcoRI) is used to cut both the plasmid vector and the human DNA containing the insulin gene.
EcoRI recognizes the palindromic sequence 5'-GAATTC-3' and cuts between the G and A on each strand, creating complementary "sticky ends" on both the plasmid and the human DNA fragments.
Step 3: Mix the Fragments and Join Them with DNA Ligase
The cut plasmid and the insulin gene fragment are mixed together.
The complementary, single-stranded "sticky ends" on the plasmid and the insert spontaneously anneal (base-pair) with each other due to hydrogen bonding.
The enzyme DNA Ligase is added. This enzyme catalyzes the formation of phosphodiester bonds, sealing the sugar-phosphate backbones of the plasmid and the insert, creating a stable, circular recombinant DNA molecule (a recombinant plasmid).
Step 4: Introduce the Recombinant DNA into Host Cells (Transformation)
The recombinant plasmids are mixed with host bacteria (e.g., E. coli) that have been treated to make their cell membranes temporarily permeable. This process is called transformation.
The bacteria take up the recombinant plasmid from the surrounding solution. Note: Not all bacteria will successfully take up a plasmid.
Step 5: Identify and Select Successful Host Cells
Because the transformation process is inefficient, the next step is to find the few bacteria that contain the recombinant plasmid.
This is done using the selectable marker on the plasmid.
The bacteria are spread onto agar plates containing the antibiotic (e.g., Ampicillin).
Selection: Only bacteria that have successfully taken up the plasmid (and thus carry the antibiotic resistance gene) will survive and form colonies. Bacteria without a plasmid will die.
Step 6: Screen for Clones with the Correct Insert
The surviving colonies on the antibiotic plate all contain a plasmid, but not necessarily the recombinant plasmid with the insulin gene insert. Some plasmids may have re-ligated without an insert.
Screening is performed to identify these "correct" clones. A common method is to use the fact that the insertion of the DNA fragment disrupts a gene in the plasmid's MCS (often the LacZ gene). Colonies with the correct insert will appear white, while those without an insert will appear blue on a special indicator plate. Alternatively, PCR or DNA sequencing can be used for confirmation.
Step 7: Grow the Selected Bacteria and Harvest the Product
A single bacterial colony containing the correct recombinant plasmid is picked and grown in a large culture broth.
As the bacteria divide, they replicate the recombinant plasmid and express the gene it carries, producing the protein of interest (e.g., human insulin).
The bacteria are then harvested, and the insulin protein is purified for medical use.
Summary: The Key Tools and Their Roles
Tool/Component | Role in Creating Recombinant DNA |
|---|---|
Restriction Enzymes | Molecular Scissors: Cut the vector and insert DNA at specific sites to create compatible ends. |
DNA Ligase | Molecular Glue: Covalently joins the DNA backbone of the vector and insert. |
Vector (Plasmid) | Molecular Vehicle: Carries the DNA insert into the host cell and provides the machinery for replication. |
Host Cell (E. coli) | Molecular Factory: Amplifies the recombinant DNA and expresses the encoded protein. |
Selectable Marker | Molecular Filter: Allows for the easy identification of host cells that have taken up the vector. |
In essence, making recombinant DNA is a molecular cut-and-paste operation. We cut a gene of interest and a plasmid vector with the same enzymatic scissors, paste them together with enzymatic glue, and then put this new construct into a cellular "copy machine" that also functions as a protein factory. This technology is the backbone of the biotech industry, enabling the production of life-saving drugs, genetically modified organisms, and advanced research tools.
what is recombinant DNA?
Creating recombinant DNA is also called gene cloning. The process involves combining DNA molecules from two different sources to create a new, hybrid DNA molecule.
The core idea is to use two key tools:
Restriction Enzymes to cut DNA at specific sites.
DNA Ligase to paste DNA fragments together.
DNA: deoxyribonucleic acid.
"Ligase": "An enzyme that binds or ties things together."
Here is a step-by-step explanation of how recombinant DNA is made, using the classic example of inserting a human gene into a bacterial plasmid to produce a protein like insulin.
Describe the effect of the following on electrophoresis: voltage gradient, current, gel concentration, charge density, pH.
Gel electrophoresis separates DNA, RNA, or proteins based on their SIZE AND CHARGE by forcing them to migrate through a gel under an electric field. Each of the parameters you listed has a distinct and crucial effect on this process.
Here’s a detailed breakdown of their effects:
1. Voltage Gradient (Volts per cm, V/cm)
What it is: The strength of the electric field applied across the gel. It's calculated as the total voltage divided by the length of the gel.
Effect:
Higher Voltage Gradient: Increases the force acting on the molecules, causing them to migrate faster. This is useful for quick separations.
Lower Voltage Gradient: Decreases the force, causing slower migration.
Trade-offs and Consequences:
Resolution: While high voltage is fast, it can generate excessive heat. This heat can melt the gel, cause bands to smear ("smiling" effect due to uneven temperature), and denature the molecules. It can also outpace the gel's sieving ability, reducing resolution between similarly sized fragments.
Optimal Setting: A moderate voltage gradient is typically used to balance speed with high resolution. For large DNA fragments, a lower voltage is often better for clear separation.
2. Current (Amperes, Amps)
What it is: The rate of electron flow through the system. In the context of electrophoresis, the charged molecules being separated (ions in the buffer) are the primary charge carriers.
Effect:
Constant Current vs. Constant Voltage: This is a critical distinction in power supply settings.
Constant Voltage: The voltage is fixed. As the run proceeds and the buffer ion concentration changes, the current will decrease over time. This is the most common and generally preferred mode.
Constant Current: The current is fixed. To maintain a constant current as resistance in the system changes, the power supply will increase the voltage over time. This can lead to a runaway heating problem if not carefully monitored.
Consequence:
Heat Production: Heat generation is proportional to the square of the current (I²R, where R is resistance). A high current produces a lot of heat, with the same negative effects (smiling, smearing) as a high voltage gradient.
3. Gel Concentration (% Agarose or % Polyacrylamide)
What it is: The density of the gel matrix, which creates a molecular sieve.
Effect:
Higher Gel Concentration: Creates a tighter, denser meshwork. This slows down all molecules and provides better resolution for smaller molecules. Smaller fragments are separated more effectively.
Lower Gel Concentration: Creates a looser, more porous meshwork. This allows larger molecules to migrate more freely and be resolved better.
Analogy: Imagine sifting rocks through a sieve.
A fine sieve (high % gel) is great for separating sand from pebbles (small fragments).
A coarse sieve (low % gel) is needed to separate large rocks (big fragments).
Standard Practice: A 1% agarose gel is common for separating DNA fragments in the 0.5-10 kb range, while a 12% polyacrylamide gel is used for resolving very small DNA fragments or proteins.
4. Charge Density
What it is: The amount of charge per unit mass of the molecule.
Effect:
DNA/RNA: The sugar-phosphate backbone gives DNA and RNA a uniform, high negative charge density that is directly proportional to their length. This is why agarose gel electrophoresis separates them purely by size—all fragments have the same charge-to-mass ratio and experience the same force, so the gel sieve alone determines their speed.
Proteins: Proteins have different and non-uniform charge densities depending on their amino acid composition. To separate proteins by size alone, they must first be denatured with a detergent like SDS (Sodium Dodecyl Sulfate), which coats the proteins and gives them a uniform negative charge density, masking their native charge.
5. pH
What it is: The acidity or alkalinity of the running buffer.
Effect:
Buffer Integrity: The pH is carefully chosen to maintain a stable chemical environment and keep the molecules intact and in their intended state.
Charge of the Molecule:
DNA/RNA: The phosphate groups are negatively charged across a wide pH range, so DNA separation is relatively insensitive to pH changes within a standard buffer (like TAE or TBE at ~pH 8.0).
Proteins (Native Gels): This is where pH is critical. The net charge of a protein depends on the pH relative to its isoelectric point (pI).
At a pH below its pI, a protein will be positively charged.
At a pH above its pI, a protein will be negatively charged.
By adjusting the buffer pH, you can control the direction and speed of a protein's migration in a native gel (without SDS).
Summary Table
Parameter | Effect on Electrophoresis | Key Consideration |
|---|---|---|
Voltage Gradient | Higher = Faster migration. Lower = Slower migration. | High voltage causes heat, leading to smearing and poor resolution. |
Current | High current causes excessive heat. | Use Constant Voltage mode for stable, predictable runs. |
Gel Concentration | Higher % = Better resolution of small fragments. Lower % = Better resolution of large fragments. | Choose the gel percentage based on the size range of your molecules. |
Charge Density | Determines the force-to-mass ratio. Must be uniform to separate by size alone. | DNA: Naturally uniform. Proteins: Require SDS to achieve uniform charge density. |
pH | Affects the net charge of the molecule, especially proteins. |
what does gel electrophoresis do?
Gel electrophoresis separates DNA, RNA, or proteins based on their SIZE AND CHARGE by forcing them to migrate through a gel under an electric field. Each of the parameters you listed has a distinct and crucial effect on this process.
voltage gradient for gel electrophoresis
high voltage gradient
lower voltage gradient
1. Voltage Gradient (Volts per cm, V/cm)
The strength of the electric field applied across the gel. It's calculated as the total voltage divided by the length of the gel.
Effect:
Higher Voltage Gradient: Increases the force acting on the molecules, causing them to migrate faster. This is useful for quick separations.
Lower Voltage Gradient: Decreases the force, causing slower migration.
Trade-offs and Consequences:
Resolution: While high voltage is fast, it can generate excessive heat. This heat can melt the gel, cause bands to smear ("smiling" effect due to uneven temperature), and denature the molecules. It can also outpace the gel's sieving ability, reducing resolution between similarly sized fragments.
Optimal Setting: A moderate voltage gradient is typically used to balance speed with high resolution. For large DNA fragments, a lower voltage is often better for clear separation.
current for gel electrophoresis
constant current vs constant voltage
2. Current (Amperes, Amps)
The rate of electron flow through the system. In the context of electrophoresis, the charged molecules being separated (ions in the buffer) are the primary charge carriers.
Effect:
Constant Current vs. Constant Voltage: This is a critical distinction in power supply settings.
Constant Voltage: The voltage is fixed. As the run proceeds and the buffer ion concentration changes, the current will decrease over time. This is the most common and generally preferred mode.
Constant Current: The current is fixed. To maintain a constant current as resistance in the system changes, the power supply will increase the voltage over time. This can lead to a runaway heating problem if not carefully monitored.
Consequence:
Heat Production: Heat generation is proportional to the square of the current (I²R, where R is resistance). A high current produces a lot of heat, with the same negative effects (smiling, smearing) as a high voltage gradient.
gel concentration
larger molecules
smaller molecules
3. Gel Concentration (% Agarose or % Polyacrylamide)
What it is: The density of the gel matrix, which creates a molecular sieve.
Effect:
Higher Gel Concentration: Creates a tighter, denser meshwork. This slows down all molecules and provides better resolution for smaller molecules. Smaller fragments are separated more effectively.
higher concentration gel→ tighter meshwork → slows down molecules and PROVIDES BETTER RESOLUTION for SMALLER MOLECULES
Lower Gel Concentration: Creates a looser, more porous meshwork. This allows larger molecules to migrate more freely and be resolved better.
lower concentration gel → looser meshwork → allows larger molecules to migrate more freely→ better resolution for LARGER MOLECULES.
Analogy: Imagine sifting rocks through a sieve.
A fine sieve (high % gel) is great for separating sand from pebbles (small fragments).
A coarse sieve (low % gel) is needed to separate large rocks (big fragments).
Standard Practice: A 1% agarose gel is common for separating DNA fragments in the 0.5-10 kb range, while a 12% polyacrylamide gel is used for resolving very small DNA fragments or proteins.
charge density
effect of charge density on DNA/RNA
effect of charge density on proteins.
4. Charge Density
The amount of charge per unit mass of the molecule.
charge density refers to the molecule, not the gel.
Effect:
DNA/RNA: The sugar-phosphate backbone gives DNA and RNA a uniform, high negative charge density that is directly proportional to their length. This is why agarose gel electrophoresis separates them purely by size—all fragments have the same charge-to-mass ratio and experience the same force, so the gel sieve alone determines their speed.
Proteins: Proteins have different and non-uniform charge densities depending on their amino acid composition. To separate proteins by size alone, they must first be denatured with a detergent like SDS (Sodium Dodecyl Sulfate), which coats the proteins and gives them a uniform negative charge density, masking their native charge.
pH
on which macromolecule does pH become critical because it does not a uniform negative charge?
5. pH: The acidity or alkalinity of the running buffer.
Effect:
Buffer Integrity: The pH is carefully chosen to maintain a stable chemical environment and keep the molecules intact and in their intended state.
Charge of the Molecule:
DNA/RNA: The phosphate groups are negatively charged across a wide pH range, so DNA separation is relatively insensitive to pH changes within a standard buffer (like TAE or TBE at ~pH 8.0).
Proteins (Native Gels): This is where pH is critical. The net charge of a protein depends on the pH relative to its isoelectric point (pI).
At a pH below its pI, a protein will be positively charged.
At a pH above its pI, a protein will be negatively charged.
By adjusting the buffer pH, you can control the direction and speed of a protein's migration in a native gel (without SDS).
Enunciate the major features of the different types of cloning vectors and their use.
Cloning vectors are DNA molecules used as vehicles to artificially carry foreign genetic material into a host cell, where it can be replicated and/or expressed. The choice of vector depends critically on the size of the DNA insert and the ultimate application (e.g., simple amplification, expression, genomic mapping).
Here is a breakdown of the major features and uses of the primary types of cloning vectors.
1. Plasmid Vectors
These are the most versatile and commonly used vectors, especially for routine cloning in E. coli.
Major Features:
Origin of Replication (ORI): Allows the plasmid to replicate independently within the bacterial host.
Multiple Cloning Site (MCS): A short DNA sequence containing recognition sites for many different restriction enzymes, making it easy to insert a DNA fragment.
Selectable Marker: A gene (e.g., for antibiotic resistance) that allows selection of host cells that have taken up the plasmid.
High Copy Number: Many plasmids are engineered to replicate into hundreds of copies per cell, enabling high-yield DNA production.
Primary Use:
General Cloning: Amplifying DNA fragments up to ~10-15 kbp.
Subcloning: Moving DNA fragments from one vector to another.
Protein Expression: Specialized expression plasmids contain strong promoters to drive high-level production of a protein of interest.
2. Phage Vectors (Bacteriophage Vectors)
These vectors are based on bacterial viruses, most famously bacteriophage lambda (λ).
Major Features:
Viral Packaging System: The recombinant DNA is packaged into a phage particle, which then infects the host bacterium with high efficiency—much higher than plasmid transformation.
Size Selection: The phage genome has "stuffer" regions that can be removed and replaced with the foreign insert. The viral capsid can only package DNA of a specific size range, automatically selecting for vectors that contain an insert.
Primary Use:
Construction of Genomic DNA Libraries: Ideal for cloning larger DNA fragments (up to ~25 kbp) because they can accommodate more DNA than plasmids.
3. Cosmid Vectors
Cosmids are hybrids that combine the features of plasmid and phage vectors.
Major Features:
Plasmid Origin of Replication (ORI) and Selectable Marker: They replicate in the host cell like a plasmid once inside.
cos Sites: They contain the cos (cohesive end) sites from bacteriophage λ, which are necessary for the phage's packaging machinery.
Large Insert Capacity: Because most of the viral genes are removed, cosmids can carry very large foreign DNA inserts (up to ~45 kbp).
Primary Use:
Cloning Large DNA Fragments, larger than what plasmids or phage vectors can handle. They were crucial for early genome mapping projects.
4. Bacterial Artificial Chromosomes (BACs)
BACs are designed to behave like a natural, stable chromosome in bacteria.
Major Features:
Low Copy Number: Based on the F-factor plasmid, they are maintained at just 1-2 copies per cell. This prevents recombination and makes them very stable, even with large inserts.
Very Large Insert Capacity: Can reliably clone DNA fragments of 100-300 kbp.
Sophisticated Control: Contain genes for strict copy number control and partitioning to ensure the vector is passed to daughter cells during division.
Primary Use:
Genome Sequencing Projects: The backbone of the Human Genome Project, as they can stably maintain large fragments of human DNA for sequencing and mapping.
Studying Large Genes and Gene Clusters.
5. Yeast Artificial Chromosomes (YACs)
YACs are engineered to act as a functional chromosome in yeast cells.
Major Features:
Contains Essential Eukaryotic Elements:
Centromere (CEN): Ensures proper segregation during cell division.
Telomeres (TEL): Protect the ends of the linear chromosome.
Autonomously Replicating Sequence (ARS): Functions as an origin of replication.
Largest Insert Capacity: Can clone enormous DNA fragments from 100 kbp up to 2,000 kbp (2 Mb).
Primary Use:
Mapping Complex Eukaryotic Genomes (e.g., human, mouse) as they can hold very large genomic regions.
Studying Functional Genomics in a eukaryotic environment.
Drawback: Less stable than BACs and can suffer from recombination, leading to chimeric clones (inserts from different parts of the genome joined together).
6. Expression Vectors
This is a functional class of vector (often based on plasmids) designed specifically to produce the protein encoded by the inserted DNA.
Major Features (in addition to standard plasmid features):
Strong Promoter: A powerful, regulatable DNA sequence that drives high-level transcription of the inserted gene.
Ribosome Binding Site (RBS): For efficient translation initiation in the host.
Termination Sequence: Signals the end of transcription.
Fusion Tags: Sequences that encode tags (e.g., His-tag, GST-tag) fused to the protein to simplify purification and detection.
Primary Use:
Producing large quantities of a specific protein for research, therapeutic, or industrial purposes (e.g., insulin, growth hormones).
Summary Table
Vector Type | Host | Typical Insert Size | Key Feature | Primary Use |
|---|---|---|---|---|
Plasmid | Bacteria | 0.1 - 15 kbp | Multiple Cloning Site (MCS), High Copy Number | Routine cloning, subcloning, protein expression |
Phage (λ) | Bacteria | 10 - 25 kbp | High-efficiency delivery via viral infection | Genomic DNA libraries |
Cosmid | Bacteria | 35 - 45 kbp | Hybrid; uses phage packaging & plasmid replication | Cloning larger genomic fragments |
BAC | Bacteria | 100 - 300 kbp | Low Copy Number, High Stability | Genome sequencing, large gene clusters |
YAC | Yeast | 100 - 2000 kbp | Functions as a true eukaryotic chromosome | Mapping very large genomic regions, functional genomics |
Expression Vector | Bacteria, Yeast, etc. | Varies | Strong promoter, fusion tags | High-level production of recombinant proteins |
In summary, the selection of a cloning vector is a strategic decision based on the size of the DNA fragment to be cloned and the desired outcome, ranging from simple amplification in a plasmid to the functional analysis of massive genomic regions in a YAC.
what are cloning vectors?
Cloning vectors are DNA molecules used as vehicles to artificially carry foreign genetic material into a host cell, where it can be replicated and/or expressed.
what does the choice of the vector depend on?
The choice of vector depends critically on the size of the DNA insert and the ultimate application (e.g., simple amplification, expression, genomic mapping).
plasmid cloning vector
1. Plasmid Vectors
These are the most versatile and commonly used vectors, especially for routine cloning in E. coli.
Major Features:
Origin of Replication (ORI): Allows the plasmid to replicate independently within the bacterial host.
Multiple Cloning Site (MCS): A short DNA sequence containing recognition sites for many different restriction enzymes, making it easy to insert a DNA fragment.
Selectable Marker: A gene (e.g., for antibiotic resistance) that allows selection of host cells that have taken up the plasmid.
High Copy Number: Many plasmids are engineered to replicate into hundreds of copies per cell, enabling high-yield DNA production.
Primary Use:
General Cloning: Amplifying DNA fragments up to ~10-15 kbp.
Subcloning: Moving DNA fragments from one vector to another.
Protein Expression: Specialized expression plasmids contain strong promoters to drive high-level production of a protein of interest.
(bacteriophage) phage cloning vector
what is the advantage of using a bacteriophage as a cloning vector vs plasmid?
stuffer region
2. Phage Vectors (Bacteriophage Vectors)
These vectors are based on bacterial viruses, most famously bacteriophage lambda (λ).
Major Features:
Viral Packaging System: The recombinant DNA is packaged into a phage particle, which then infects the host bacterium with high efficiency—much higher than plasmid transformation.
Size Selection: The phage genome has "stuffer" regions that can be removed and replaced with the foreign insert. The viral capsid can only package DNA of a specific size range, automatically selecting for vectors that contain an insert.
Primary Use:
Construction of Genomic DNA Libraries: Ideal for cloning larger DNA fragments (up to ~25 kbp) because they can accommodate more DNA than plasmids.
cosmid vectors
3. Cosmid Vectors
cos: “cohesive end site” the cos site is a specific DNA sequence found in the genome of bacteriophage lambda (a virus that infects bacteria). These "sticky ends" are essential for the viral DNA to be packaged into the phage protein coat.
mid: plasmid
Cosmids are hybrids that combine the features of plasmid and phage vectors.
Major Features:
Plasmid Origin of Replication (ORI) and Selectable Marker: They replicate in the host cell like a plasmid once inside.
"Replicates in the host cell" describes the outcome or the process.
The Origin of Replication is the specific location where that process begins.
cos Sites: They contain the cos (cohesive end) sites from bacteriophage λ, which are necessary for the phage's packaging machinery.
Large Insert Capacity: Because most of the viral genes are removed, cosmids can carry very large foreign DNA inserts (up to ~45 kbp).
Primary Use:
Cloning Large DNA Fragments, larger than what plasmids or phage vectors can handle. They were crucial for early genome mapping projects.
Creating genomic and cDNA libraries are fundamental techniques for isolating and studying genes. They serve different purposes: a genomic library represents the entire genome, including introns, promoters, and non-coding regions, while a cDNA library represents the expressed genes (the transcriptome) in a specific cell or tissue at a specific time.
Here is a step-by-step description of how each library is constructed.
Genomic DNA Library
The goal is to create a collection of clones that together contain every sequence from the entire genome of a species.
Step 1: Isolate and Purify Genomic DNA
Extract high-molecular-weight DNA from the cells of the target species. The DNA must be as intact as possible to allow for the cloning of large fragments.
Step 2: Fragment the Genomic DNA
This can be done in two ways:
Partial Restriction Digest: Use a restriction enzyme (e.g., Sau3A) with a 4-base pair recognition site under conditions where it only cuts a subset of its available sites. This produces a random, overlapping set of fragments essential for complete coverage.
Physical Shearing: Pass the DNA through a narrow syringe or use sound waves (sonication) to break it randomly into fragments. The ends are then repaired enzymatically to make them blunt-ended for ligation.
Step 3: Choose an Appropriate Vector
The choice depends on the size of the DNA fragments and the genome size.
Plasmids: For small genomes (e.g., bacteria) or small fragments (< 15 kb).
Bacteriophage Lambda (λ): For fragments 10-25 kb.
Cosmids: For fragments 35-45 kb.
BACs (Bacterial Artificial Chromosomes): For very large fragments (100-300 kb), essential for large eukaryotic genomes.
Step 4: Ligate DNA Fragments into the Vector
The fragmented genomic DNA is mixed with the prepared vector that has been cut with a compatible restriction enzyme.
The enzyme DNA Ligase is added to covalently join the DNA fragments into the vector molecules.
Step 5: Introduce the Recombinant Molecules into Host Cells
The ligation mixture is introduced into a host (usually E. coli) via:
Transformation (for plasmids/cosmids/BACs).
In vitro Packaging & Infection (for phage vectors, where the DNA is packaged into viral particles that then infect the bacteria).
Step 6: Collect and Store the Library
The transformed cells or phage plaques are collected and stored. This entire collection, which (ideally) contains at least one clone for every sequence in the genome, is the genomic library.
Key Consideration: To ensure a high probability that every genomic sequence is present, a library must contain a very large number of clones. This is calculated as the "number of clones needed" using a formula based on genome size and average insert size.
cDNA (Complementary DNA) Library
The goal is to create a collection of clones that represent only the coding sequences (exons) of genes that were being actively expressed in a specific cell type or tissue at the time of RNA extraction.
Step 1: Isolate mRNA from the Specific Tissue
Extract total RNA from the cells of interest.
Exploit the poly-A tail of eukaryotic mRNA by passing the total RNA over a column containing oligo(dT) cellulose. The poly-A tails bind, and everything else is washed away. The purified mRNA is then eluted.
Step 2: Synthesize First Strand of cDNA
Use the enzyme Reverse Transcriptase and a primer to start DNA synthesis.
Primer: An Oligo(dT) primer that binds to the poly-A tail of the mRNA.
This creates an mRNA-DNA hybrid molecule.
Step 3: Synthesize Second Strand of cDNA
There are several methods, but a common one is:
RNAse H Method:
The enzyme RNAse H is added, which nicks the mRNA strand in the mRNA-DNA hybrid.
The remaining mRNA fragments now serve as primers for DNA Polymerase I, which uses the first DNA strand as a template to synthesize the second DNA strand.
The result is a double-stranded cDNA molecule that accurately represents the original mRNA sequence, but without introns.
Step 4: Ligate cDNA into a Vector
The double-stranded cDNA is prepared for ligation. Short, synthetic DNA sequences called linkers or adaptors (containing a restriction site) are often attached to the ends of the cDNA fragments.
The cDNA is then ligated into a suitable plasmid or phage vector, just as in genomic library construction.
Step 5: Clone into Host Bacteria
The recombinant vectors are introduced into E. coli host cells via transformation or infection.
Step 6: Collect and Store the Library
The resulting collection of bacterial clones, each containing a cDNA copy of an mRNA from the original tissue, is the cDNA library.
Summary Table: Genomic vs. cDNA Library
Feature | Genomic Library | cDNA Library |
|---|---|---|
Source DNA | Total genomic DNA from any cell type. | mRNA from a specific cell type, tissue, or condition. |
Represents | The entire genome: coding and non-coding regions (introns, promoters, etc.). | Only the expressed (transcribed) genes; exons only. |
Introns | Present. | Absent (removed during mRNA processing). |
Complexity | Very large; number of clones depends on genome size. | Smaller; number of clones depends on the diversity of mRNA. |
Tissue/Stage Specificity | No. Same for all cells of an organism. | Yes. Represents the gene expression profile at the time of isolation. |
Primary Use | Studying gene structure, promoters, introns, genomic organization. | Studying gene expression, discovering coding sequences, producing eukaryotic proteins in bacteria (since no introns need to be spliced). |
In essence, you make a genomic library by fragmenting the entire DNA of an organism and cloning all of it. You make a cDNA library by capturing the "memo" copies of the active genes (mRNA), converting them back into stable DNA, and then cloning that subset. The choice of which library to use depends entirely on the biological question being asked.
Mention some clinical uses of recombinant DNA
Recombinant DNA technology has revolutionized modern medicine, moving from the laboratory to the clinic in countless ways. Its clinical uses are vast and can be broadly categorized as shown in the following diagram, which highlights the four main areas of application:
Here is a detailed breakdown of these clinical applications, with specific examples for each category.
1. Production of Therapeutic Proteins and Vaccines
This is the most established and widespread clinical use. Instead of extracting proteins from animal or human sources, we can now produce pure, human-specific proteins in bacteria, yeast, or mammalian cells.
Therapeutic Proteins:
Insulin (for Diabetes): The first recombinant drug, approved in 1982. Replaced pig and cow insulin, which could cause allergic reactions. Human insulin is now produced in E. coli or yeast.
Human Growth Hormone (for Growth Disorders): Replaced the previous source (extraction from human cadavers), which carried the risk of transmitting Creutzfeldt-Jakob disease. Recombinant HGH is safe and abundant.
Coagulation Factors (for Hemophilia): Recombinant Factor VIII and IX have eliminated the risk of blood-borne infections (like HIV and Hepatitis) that were a tragic consequence of pooled plasma-derived products in the past.
Erythropoietin (EPO) (for Anemia): Used to treat anemia associated with chronic kidney disease and chemotherapy.
Monoclonal Antibodies (for Cancer, Autoimmune Diseases): Drugs like Rituximab (for lymphoma) and Adalimumab (for rheumatoid arthritis, psoriasis) are recombinant antibodies that specifically target disease-causing molecules.
Vaccines:
Subunit Vaccines: Instead of using a whole killed or attenuated virus, these vaccines use a single, recombinant protein from the pathogen to generate a protective immune response.
Hepatitis B Vaccine: Composed of recombinant hepatitis B surface antigen (HBsAg) produced in yeast. It is extremely safe and effective.
HPV Vaccine (Gardasil, Cervarix): Made from recombinant virus-like particles (VLPs) of the human papillomavirus capsid protein, preventing infection and cervical cancer.
2. Advanced Diagnostics and Screening
Recombinant DNA technology provides the tools for highly sensitive and specific detection of diseases.
Infectious Disease Testing:
PCR and RT-PCR Tests: The enzymes (DNA polymerases) and reagents used in PCR are produced recombinantly. This is the gold standard for detecting pathogens like SARS-CoV-2, HIV, and Hepatitis C by amplifying their genetic material.
Rapid Antigen Tests: The antibodies used in these tests are often produced using recombinant methods.
Genetic Testing:
Carrier Screening: Identifying individuals who carry a mutation for a recessive disorder like Cystic Fibrosis or Tay-Sachs disease.
Prenatal Diagnosis: Detecting chromosomal abnormalities and genetic disorders in a fetus.
Cancer Prognostics: Tests like Oncotype DX use recombinant techniques to analyze the expression levels of multiple genes in a tumor to predict the likelihood of recurrence and guide treatment decisions.
3. Gene Therapy and Gene Editing
This is a more recent and revolutionary application, aiming to treat the root cause of genetic diseases by correcting the faulty gene itself.
Gene Therapy: Introducing a functional copy of a gene into a patient's cells to compensate for a non-functional one.
Example: Luxturna is an FDA-approved gene therapy for a specific form of inherited blindness (Leber congenital amaurosis). It uses a harmless recombinant virus (AAV) to deliver a functional copy of the RPE65 gene directly into retinal cells.
Example: Chimeric Antigen Receptor (CAR) T-cell therapy for cancer. A patient's own T-cells are genetically engineered ex vivo using recombinant viruses to express a receptor that targets their cancer, then infused back into the patient.
Gene Editing (e.g., CRISPR-Cas9): This technology itself relies on recombinant components (the Cas9 enzyme and guide RNA). It allows for precise correction of mutations within the genome.
Example: The first CRISPR-based therapies have been approved for Sickle Cell Disease and Beta-Thalassemia. They involve editing a patient's own hematopoietic stem cells to reactivate fetal hemoglobin, effectively curing the disease.
4. Personalized Medicine and Pharmacogenomics
Recombinant technology enables the development of tests that tailor medical treatment to an individual's genetic profile.
Pharmacogenomics: Determining how a patient's genes affect their response to drugs.
Example: Before prescribing the HIV drug Abacavir, patients are tested for a specific HLA allele (HLA-B*5701) using recombinant DNA-based assays. Those with the allele are at high risk for a severe hypersensitivity reaction and should not receive the drug.
Cancer Targeted Therapy:
Example: Patients with non-small cell lung cancer are tested for mutations in the EGFR gene. If the mutation is present, they can be treated with targeted drugs like Gefitinib, which are much more effective than standard chemotherapy for this specific genetic subtype.
In summary, recombinant DNA technology is the backbone of modern biotherapeutics, has created a new era of safe and effective vaccines and diagnostics, and is now unlocking the potential of gene-based medicine, where treatments are designed to target an individual's unique genetic makeup and the specific molecular cause of their disease.
we are trying to modify the genotype to get a new phenotype.

YOU WILL BE ASKED ABOUT THE STEPS OF TRANSFORMATION IN THE EXAM.
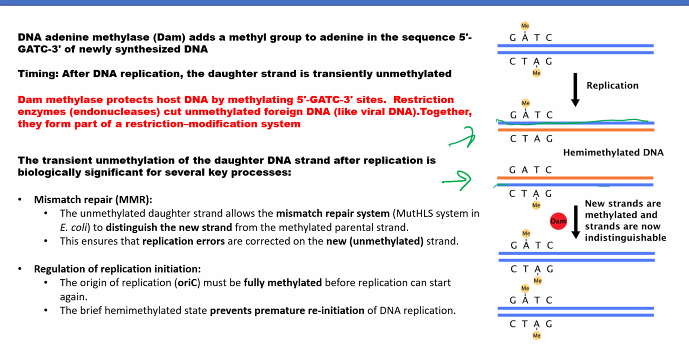
palindromes
methylation

the methylated strand is the original.
the cells use the dna methylation system and they use the strand that is not methylated.
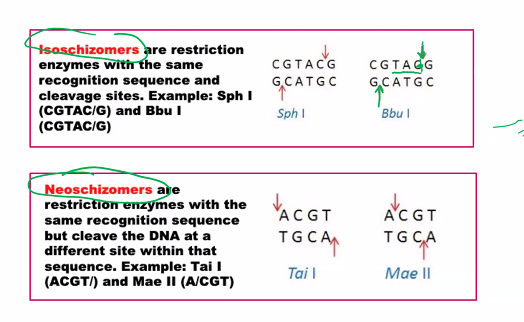
isoschizomers
neoschizomers
This is a classic topic in molecular biology, specifically in the context of restriction enzymes. The terms isoschizomers and neoschizomers describe different types of relationships between these enzymes.
The key to understanding both terms lies in their recognition of the same DNA sequence, but with a critical difference in how they cut it.
Let's break them down.
Isoschizomers
Etymology: From the Greek isos (equal) and schizein (to split).
Definition: Isoschizomers are pairs (or groups) of restriction enzymes that recognize the same specific DNA sequence AND cut at the exact same position within that sequence.
Key Characteristics:
Same Recognition Sequence: They bind to the same string of nucleotides (e.g., both recognize
G*AATTC).Same Cut Site: They produce identical cut ends. If one produces a 5' overhang, the other does too. If it's a blunt end, the other also produces a blunt end.
Example:
SphI (from Streptomyces phaeochromogenes) and BbuI (from Bacillus brevis) are isoschizomers.
Recognition Sequence for both:
G CATGC^CCut Site for both: They both cut between the G and C on the top strand (as indicated by
^) and at the symmetrical position on the bottom strand, producing the same sticky ends.
Practical Implication: Isoschizomers are often functionally interchangeable in cloning experiments. You might use one from a different supplier or one that is more affordable or has a better storage buffer.
Neoschizomers
Etymology: From the Greek neos (new) and schizein (to split).
Definition: Neoschizomers are pairs of restriction enzymes that recognize the same specific DNA sequence BUT cut at different positions within that sequence.
Key Characteristics:
Same Recognition Sequence: They bind to the same string of nucleotides.
Different Cut Sites: They cut the DNA backbone at different points, resulting in different ends.
Example (The Classic One):
SmaI and XmaI are neoschizomers.
Recognition Sequence for both:
C CC GGGCut Site for SmaI: It cuts straight through the center, producing blunt ends.
CCCGGG
Cut Site for XmaI: It cuts between the first and second cytosines, producing sticky ends.
CCCGGG
Practical Implication: Neoschizomers are NOT interchangeable. The type of ends they produce (blunt vs. sticky) is critical for determining which DNA fragments can ligate together. Using SmaI instead of XmaI would completely change the outcome of your experiment.
Summary Table
Feature | Isoschizomers | Neoschizomers |
|---|---|---|
Etymology | "Equal cutter" | "New cutter" |
Recognition Sequence | Identical | Identical |
Cut Position | Identical | Different |
Ends Produced | Identical (e.g., both 5' overhang) | Different (e.g., blunt vs. sticky) |
Functional in Lab? | Usually interchangeable | NOT interchangeable |
Analogy | Two different brands of scissors that cut paper exactly the same way. | Two tools that both recognize a "perforated line" on a coupon: one cuts the line perfectly, the other cuts 2mm to the left. |
A Third Related Term: Isocaudomers
For completeness, there's a third term: Isocaudomers (from Greek cauda, meaning "tail").
These are enzymes with different recognition sequences that, after cutting, produce the same sticky ends.
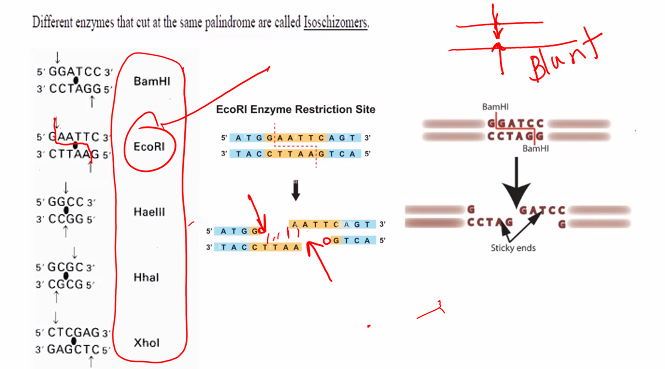
Example: BamHI (G^GATCC) and BglII (A^GATCT) are isocaudomers. They both produce the same 5' GATC overhang, allowing fragments generated by one enzyme to be ligated to fragments generated by the other. This was very useful in early cloning strategies.
what are the two types of ends?
sticky ends and blunt ends.
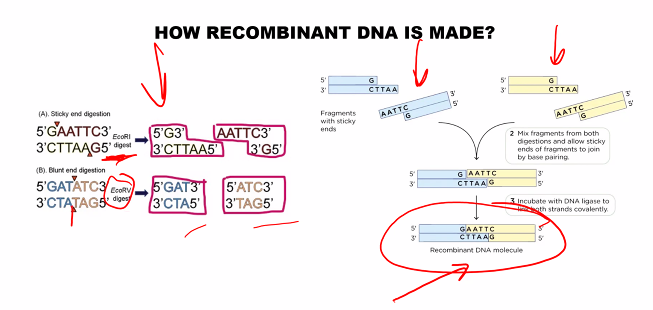
recombination dna meaning and mechanism
palindrome
AND
why is the correct answer to the question D?
A palindrome is a word, phrase, number, or other sequence of characters that reads the same forward and backward (ignoring spaces, punctuation, and capitalization).
2. Molecular Biology Meaning (Crucial for DNA)
In molecular biology, a palindrome or palindromic sequence refers to a specific kind of symmetry in a DNA (or RNA) sequence.
A DNA sequence is palindromic if the sequence of its nucleotides is the same on one strand as it is on the complementary strand when both are read in the same 5' to 3' direction.
This is often described as having twofold rotational symmetry.
How to Visualize It:
Imagine a double-stranded DNA segment. The sequence on the top strand, read from 5' to 3', is identical to the sequence on the bottom strand, read from 5' to 3'.
Example of a Short Palindromic Sequence:
text
5' - G A A T T C - 3'
3' - C T T A A G - 5'Read the top strand from left to right (5'→3'):
G-A-A-T-T-CRead the bottom strand from left to right (5'→3'):
G-A-A-T-T-C
They are identical!
Why are Palindromic Sequences So Important in Genetics?
Recognition Sites for Restriction Enzymes:
The vast majority of Type II restriction enzymes (molecular "scissors" used in DNA cloning) recognize and cut within short palindromic sequences (typically 4-8 base pairs long).
The symmetry of the site means the enzyme, which often functions as a homodimer (two identical subunits), can bind to the DNA in the same way on each strand.
Protein Binding Sites:
Many transcription factors (proteins that control gene expression) bind as dimers to palindromic or partially palindromic sequences in gene promoters. The symmetry of the DNA site matches the symmetry of the protein dimer.
Cruciform Structures:
Under certain conditions (e.g., negative supercoiling), a palindromic sequence can fold back on itself to form a cruciform structure (shaped like a cross). This happens when the two strands base-pair with themselves instead of with each other.
Summary
Feature | Linguistic Palindrome | DNA Palindrome |
|---|---|---|
Core Idea | Reads the same forwards and backwards. | The sequence is identical on both strands when read in the same 5'→3' direction. |
Example | "racecar" |
|
Key Importance | Wordplay, puzzles. | Restriction enzyme cutting, transcription factor binding, DNA structure. |
In essence, the palindromic sequence in DNA is a fundamental form of symmetry that is exploited by countless proteins to recognize and interact with the double helix in a specific and predictable way. It is one of the cornerstones of genetic engineering.

plasmids
C)

D)
plasmids do not provide a source of energy.


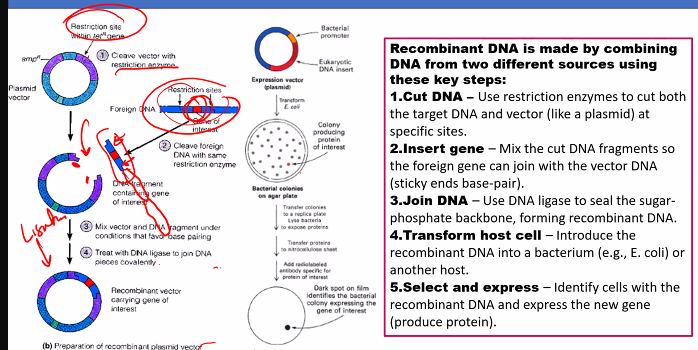

exam question!

D)



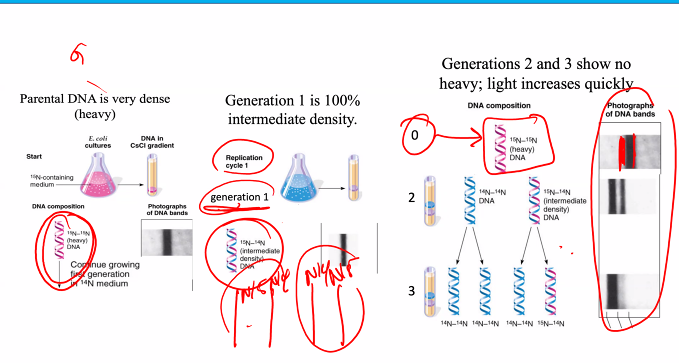
there was a picture that you missed it.


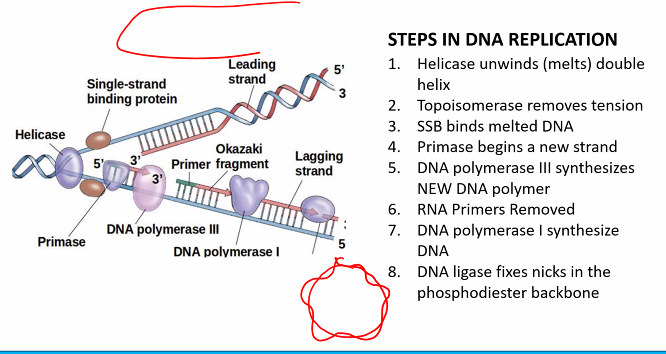
the drawing means nothing
3-OH
RNA primase (doesn’t need 3-OH)
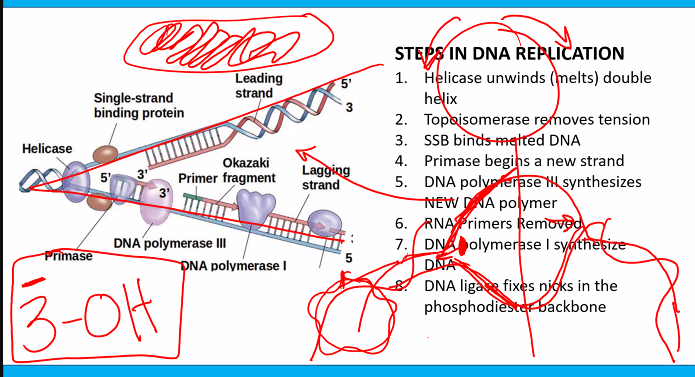
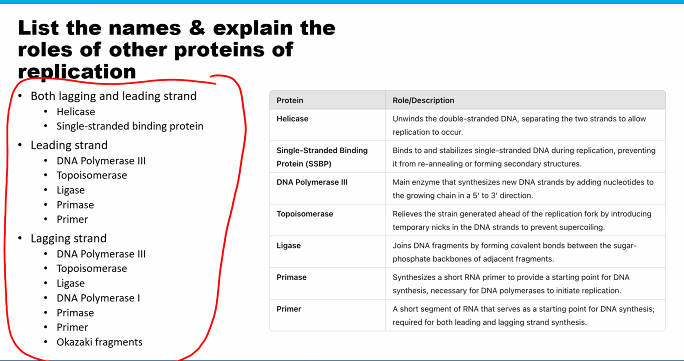
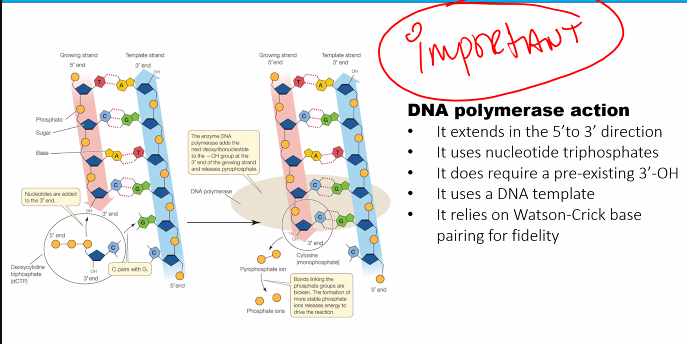
this can be used for the exam question "which of the following is not a characteristic of the DNA polymerase”?
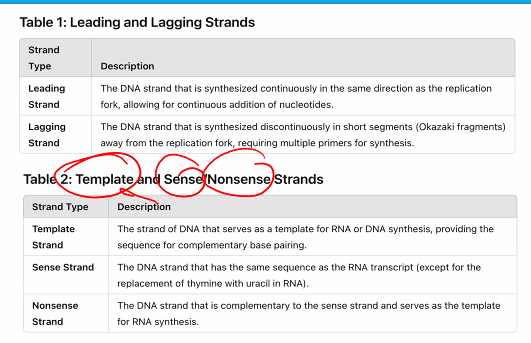
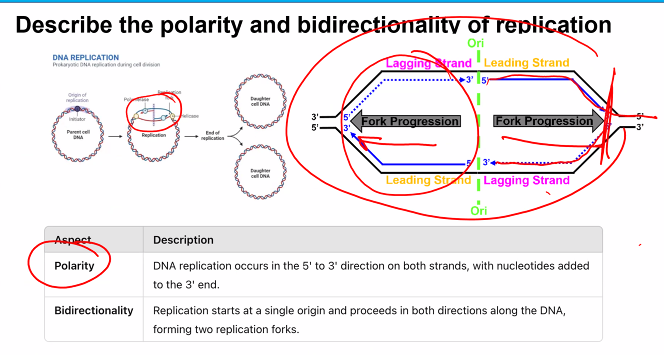
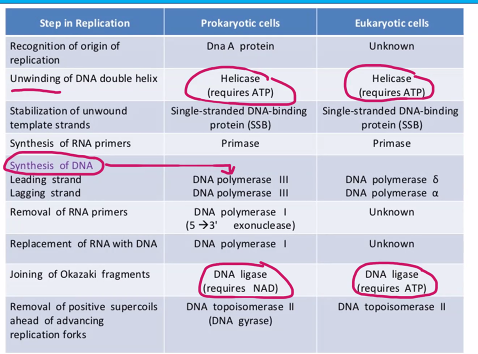
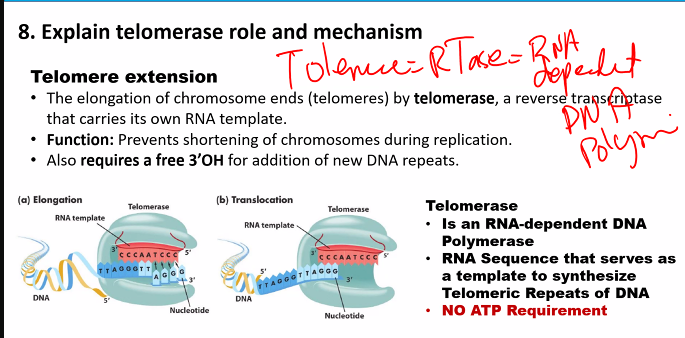


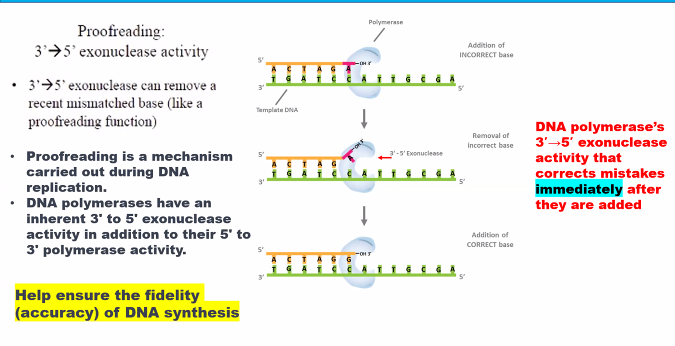
D)

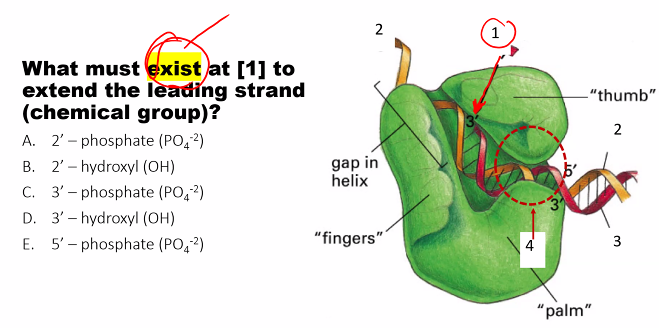
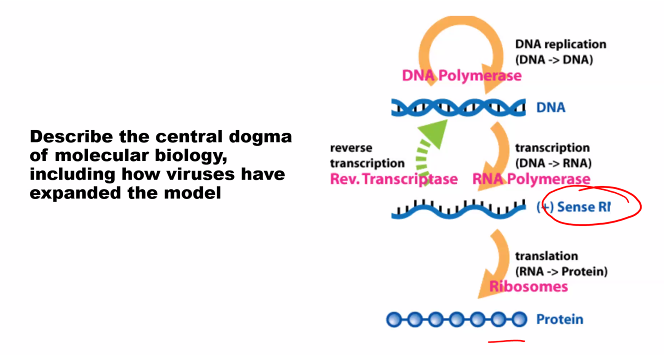
need to know the name of the subunits and their function.

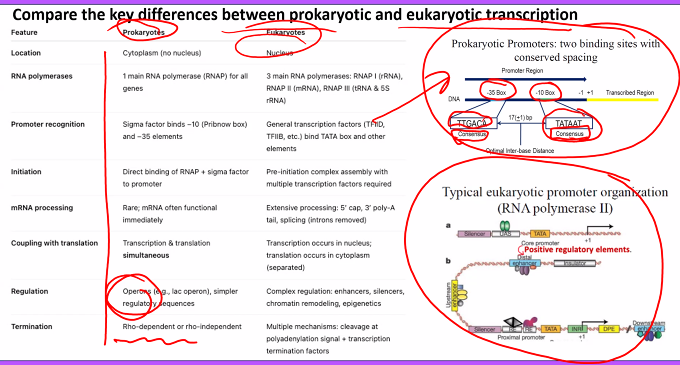
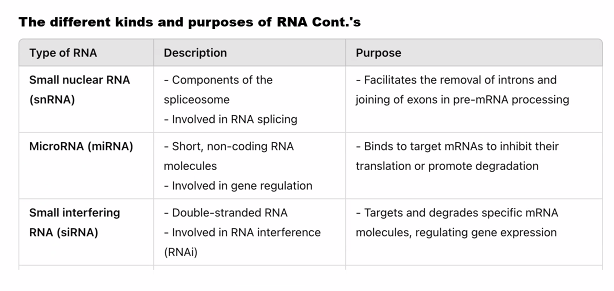
C)
methylation SILENTS the gene.

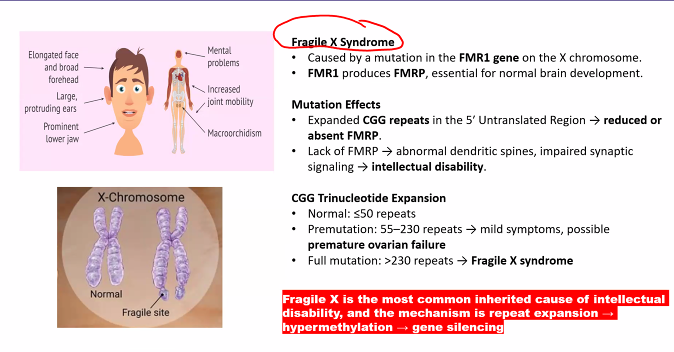
a)