LP2.11 Plasmid expansion and verification
1/48
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
49 Terms
What checks should we do when assessing our plasmid verification Sanger sequencing results?
1. Check for peak clarity - evenly spaced and single colour peaks with minimal baseline noise (variation in peak heights of up to 3 fold is normal)
2. Check automated base calling - gaps may be slightly larger between bases G-A leading to the computer believing there is a base in the gap with a low read and thus adding an "N" between them
3. Check for deterioration of signal - Decreasing signal quality throughout the read may indicate too much template present which interferes with the reaction. (A slow decrease in resolution is normal, this is worrying if it happens early)
What is PCR?
Polymerase chain reaction (PCR) is a technique that is used to exponentially amplify a specific target sequence of DNA.
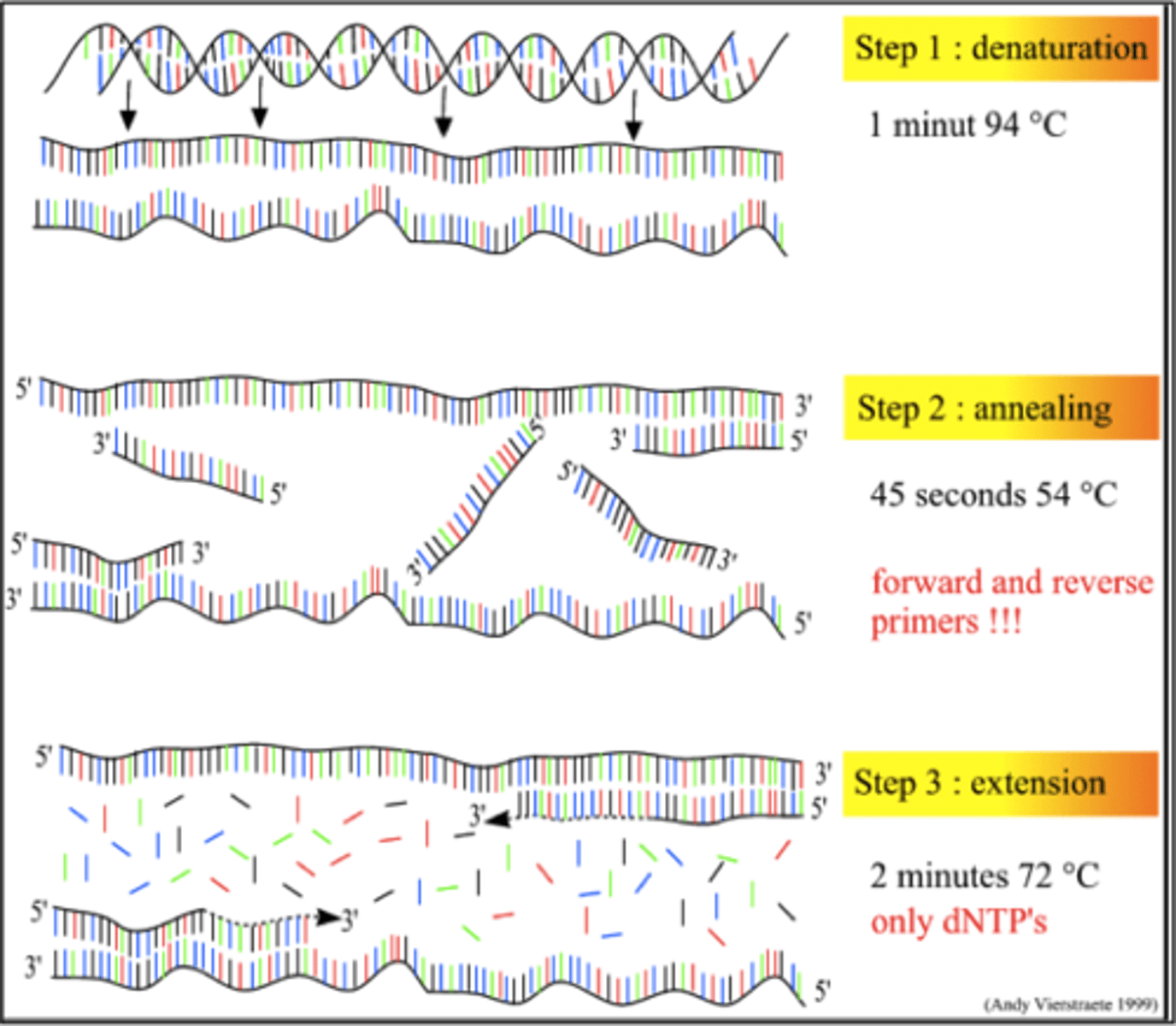
What components have to be in the solution for PCR?
Target RNA
Primers
Nucleotides
DNA polymerase
Buffer
MgCl
What are the steps of PCR?
1. Denaturation
2. Primer annealing
3. DNA polymerase catalysed DNA polymerase
This happens multiple times creating exponential quantities of the target DNA.
What are we using PCR for in our experiment and how does this work?
Plasmid verification
Confirming that our plasmid has our gRNA sequence in it.
This works because the solution contains a U6 forward primer and a gRNA specific reverse oligo, this results in a PCR product only if the gRNA is present in the plasmid.
The U6 promoter is upstream of the Bbsl restriction and gRNA insertion sites
Why do we not perform expansion before PCR since we cannot PCR bacteria?
This would be very time consuming and the denaturation step should lead to the plasmid being released into the PCR mix as it is. This is known as a colony PCR or a "dirty" PCR as the template is not pure.
Why do we test each colony serpeately?
More probability of picking one that has worked and contains a gRNA-annealed plasmid.
What does a colony PCR water negative control allow?
It rules out the possibility of a false positive as a band in a water only control should be impossible. If there is a band in your negative control the same band is likely to be in all your samples (assuming you use the same source of water).
What are the 5 elements we can troubleshoot for colony PCR?
1 - Melting and annealing temperature
2 - Cycle number
3 - Amount of DNA template
4 - Extension time
5 - Reagents and their concentrations
What is a meting temperature and what does it tell us?
Melting temperature (Tm) is defined as the temperature at which half the molecules of DNA in a solution becomes single stranded. This give us an indication of how stable the DNA is as a double stranded molecule—it gives us an idea of how much energy is required to disrupt the base pairing.
What is the equation for Tm (melting temperature)?
Tm = 2(A+T) + 4(G+C)
(estimate that does not account for salts in buffers, pH or primer concentration_
Why is melting temperature more affected by GC bonds than AT?
3 hydrogen bonds between C and G
2 hydrogen bonds between A and T
What can Tm tell us about optimising our PCR reaction?
Tm of our primers can help us decide the annealing temperature (Ta).
Most primers with temperature between 52-58 work best and this with over 65 often form secondary structures, which will impair their ability to bind to the template DNA.
Pairs of primers with very different Tm can be difficult to optimise as they bind efficiently at different temperatures.
A NEB Tm calculator can be used to figure out annealing temperatures.
What is optimal annealing temperature for a colony PCR?
A few degrees below lowest melting temperature.
What can you do if your primers have very different Tms?
Touchdown PCR
We start at a higher annealing temperature, for example 65⁰C, for the first PCR cycle. We then reduce this down by one degree for each following cycle, until we reach our target annealing temperature, for example 55⁰C. This allows each primer to bind at it's optimal temperature.
When should you do a touchdown PCR?
You should do this if the difference in Tm of the primers is greater than 2 degrees
What are the disadvantages of a touchdown PCR?
Greater non-specific binding at lower temperatures
There will be more of the products that have been copied at the higher temperature
Why could increasing cycle number not necessarily lead to more product in a colony PCR?
The reaction will plateau due to the exhaustion of reagents.
What can we do if we get non-specific bands?
Increase the amount of DNA template as too little template can mean poor amplification but using too much can lead to reduced specificity
How can we vary extension time in a colony PCR?
The length of the extension time varies according to the size of the amplicon. Too long an extension time might cause unwanted products!
If multiple bands are detected try reducing extension time.
How can GC content affect colony PCR?
GC content affects primer melting temperature, but also the efficiency of the Taq polymerase. Sometimes a different Taq or a different buffer must be used for GC rich (>65%) regions.
Why is magnesium included in colony PCR?
Magnesium is a cofactor of Taq DNA polymerase. Too little and the Taq will not function; too much and the Taq will not work specifically enough and be over active. 1mM to 5mM tends to work well.
How may too many or too few dNTPs affect a PCR?
Too many - low specificity
Too few - low efficiency
Where may you see a primer dimer and does this make the experiment void?
No, this does not but may indicate less than optimum primer to template ratio leading to primers annealing to one another. This is also common if annealing temperature is too low as this increases non-specific binding. They are typically seen at 50bp or less.
How do we run and observe the products of a colony PCR?
Run the products on an agarose gel.
How can we find out the size of our primer?
Perform 'virtual cloning' and 'virtual PCR' experiments using online tools, including Benchling, to digitally predict the size of our product
What does the intensity of the band in a colony PCR represent?
How much bacteria was picked off the plate
What do bands above the predicted PCR band suggest?
This could be:
- double insert (insert incorporated twice into plasmid)
What do bands below the predicted PCR band suggest?
This could be:
- primer dimers
- "empty" plasmid not containing your insert
What does high PCR background noise suggest?
Many non-specific bands may be a sign of contamination
Why do we need to expand our bacterial colonies?
In order to extract sufficient quantities of plasmid for mammalian transfection
Why do we add antibiotic to our culture in Step 1 of the expansion protocol?
To select for bacteria with our chosen plasmid that carries antibiotic resistance. If our plasmid isn't present then the bacteria are more likely do die in the presence of antibiotic.
Why do we set up cultures for expansion first thing in the morning?
It maximises growth time for your starter cultures before setting up the maxicultures at the end of the lab session.
Why do we use separate tubes for each gRNA (expansion)?
If we mix the gRNA plasmids at this stage one may outcompete the others resulting in you only expanding one of your gRNA plasmids. By doing separately you should get high yields of all 3 gRNAs. You can combine them much later once you are at the transfection stage.
Click to flip
Why can we not do a general lysis of the bacterial cells to purify our plasmid?
We would extract all the extrachromosomal bacterial DNA with very impure plasmid.
How do we extract pure plasmid from our bacterial cells?
Alkaline lysis:
1: Lysing cells under alkaline conditions: denatures all DNA in the cell (both genomic and plasmid)
2: Neutralisation: allows small sections of plasmid DNA to renature to dsDNA whilst the larger genomic DNA will not
Why do we not want the bacteria stored in the 4 degree fridge longer than necessary?
They may lyse on their own. Controlling lysis is essential to plasmid extraction.
Why is it essential the genomic DNA is not damaged in the process of lysis?
Shorter DNA fragments re-anneal, longer ones do not.
Which kit will we use for our 200mL bacterial culture expansion?
Maxiprep kit (Qiagen)
How can we perform a plasmid concentration?
Using the Nanodrop. If the concentration is too low it can be increased using the Speedvac concentrator which evaporates some of the liquid and thus increases the plasmid concentration. This must be done with OPEN lids.
After this you will need to resuspend in a new volume of liquid.
Define Sanger Sequencing
Sanger sequencing is a chain termination DNA sequencing method which we will be use to check the construction of our recombinant plasmid. This is a PCR based method.
What are the components of a Sanger sequencing?
- Primers binding upstream of the region of interest
- DNA polymerase
- Deoxynucleotides (dNTPs)
- Dideoxynucleosides (ddNTPs (lacking 3'OH for chain termination))
- Fluorescent markers (on the ddNTPs for detection)
What colour are the fluorescent tags on the ddNTPs?
black for guanine
green for adenine
blue for cytosine
red for thymine
These allow us to see at which
How are DNA fragments separated in Sanger sequencing?
DNA fragments are separated by size using capillary electrophoresis.
Describe the process of capillary electrophoresis
The PCR products are injected into a long glass capillary filled with a gel polymer and a cathode at one end and an anode at the other. On application of an electrical field, the negatively charged DNA fragments move towards the positive cathode at a speed determined by the size of the fragment.
At the end of the capillary, there is a window containing a laser that excites the ddNTP's fluorescent marker. The fluorescent marker then emits light at a specific wavelength that is detected by a light sensor. Software can then interpret this signal and convert it to a "base call".
What is the resolution of capillary electrophoresis?
1 base pair
How is capillary electrophoresis data presented?
Electropherogram where each coloured peak corresponds to a specific base in the sequence.
Once you have your results from Sanger sequencing, how can you compare it to your gRNA expected sequence?
Insert the data into Benchling with both sequences
What are the main features of a figure legend?
Heading
Method
Results
Definitions/Key/Descriptions