Amino Acid Metabolism - Nitrogen Economy and Urea Cycle
1/65
Earn XP
Description and Tags
Includes information from all slides; does not include main urea cycle pathway
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
66 Terms
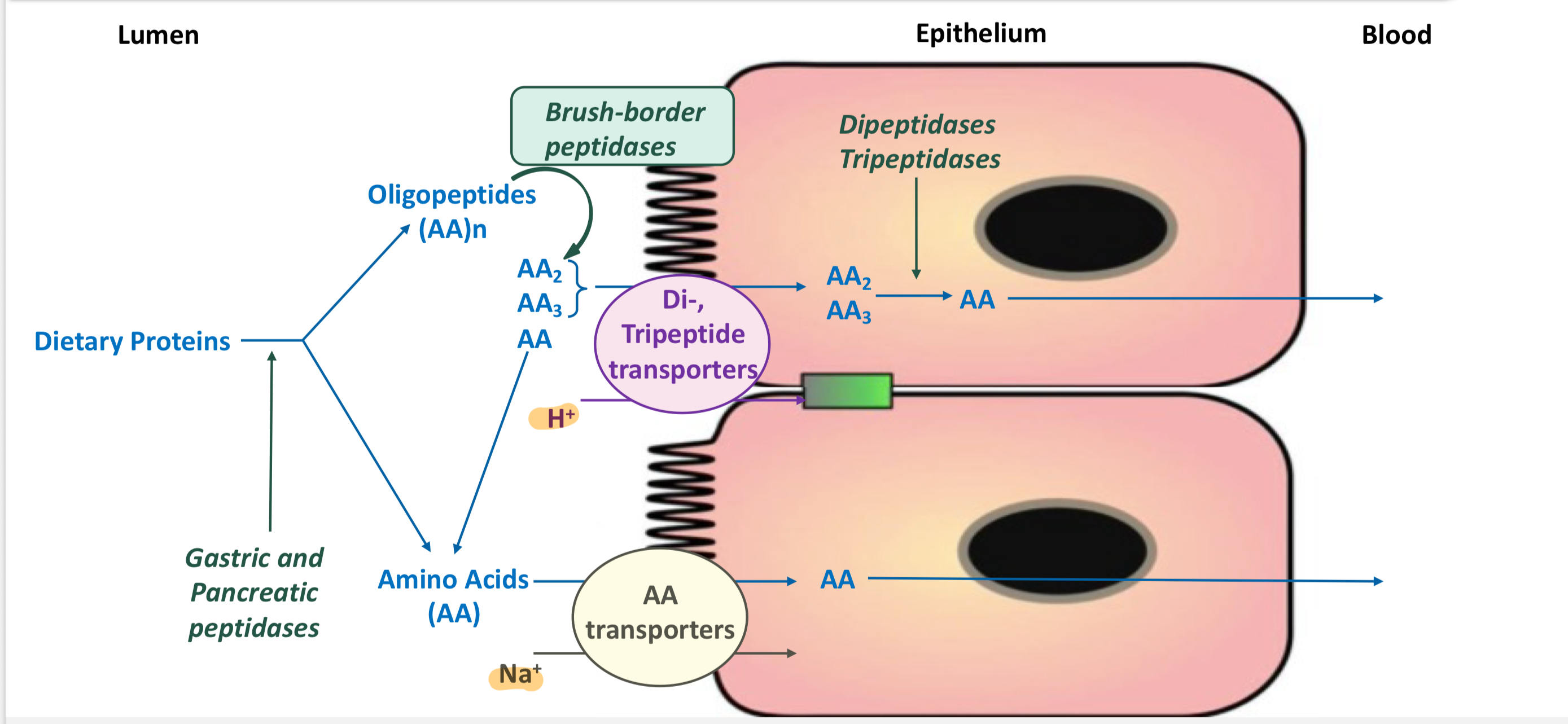
Dietary proteins are digested to absorbable forms, known as…
amino acids, dipeptides, and tripeptides
What is the exception to protein digestion?
Antibodies in maternal milk are taken up intact by infants
Where does digestion of dietary proteins begin?
In the stomach. Proteins are the only macromolecule whose digestion begins in the stomach.
What are the main enzymes involved in digestion of proteins in the stomach?
Pepsin and rennin
Pepsin is found in adult stomach, while rennin is present in infants
Pepsin - secretion, optimum pH, products, type of enzyme
Pepsin, an endonuclease, is secreted by chief cells in the inactive form - pepsinogen - which is activated by HCl (which is secreted by parietal cells). Pepsinogen can also be activated auto-catalytically by another pepsin molecule. pH optimum is 2.0 to 3.0. It produces peptides and a few amino acids.
Rennin - secretion, optimum pH, products, type of enzyme
Rennin is present in infants and secreted as pro-rennin. pH optimum is 4.0. Acts on casein of milk, which is involved in the curdling of milk.
Hyperchlorhydria
Increased HCl levels due to increased gastrin production
Achlorhydria/hypochlorhydria
Decreased HCl levels, due to pernicious anemia - autoimmune destruction of parietal cells; antacids; gastric bypass procedures, etc)
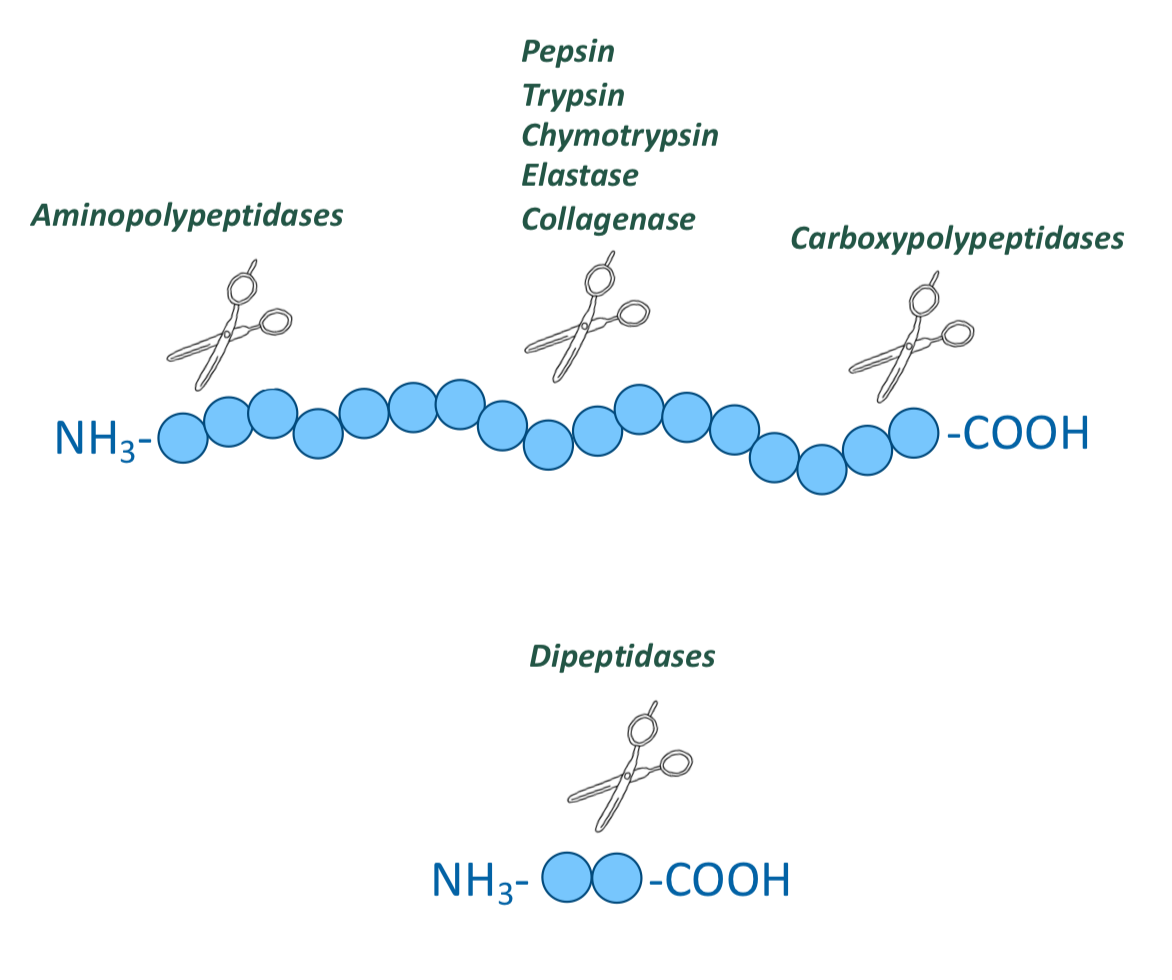
Digestion in the small intestine (duodenum) is done by which enzymes
pancreatic proteases (specific for different side chain groups of AA), endopeptidases, and exopeptidases
What are the endopeptidases involved in digestion of proteins in the small intestine?
Trypsin, chymotrypsin, elastase, and collagenase
What are the exopeptidases involved in digestion of proteins in the small intestine?
Carboxypolypeptidases A and B
What are the pro-enzymes/zymogens of the enzymes involved in digestion in the small intestine?
Trypsinogen → trypsin (activated by intestinal brush-border enzyme enterokinase)
chymotrypsinogen → chymotrypsin
Procarboxypeptidases A and B → carboxypeptidases A and B
Proelastase → elastase
Rest of the zymogens are activated by trypsin
Release and activation of proteases from small intestine is mediated by
cholecystokinin and secretin - GI hormones
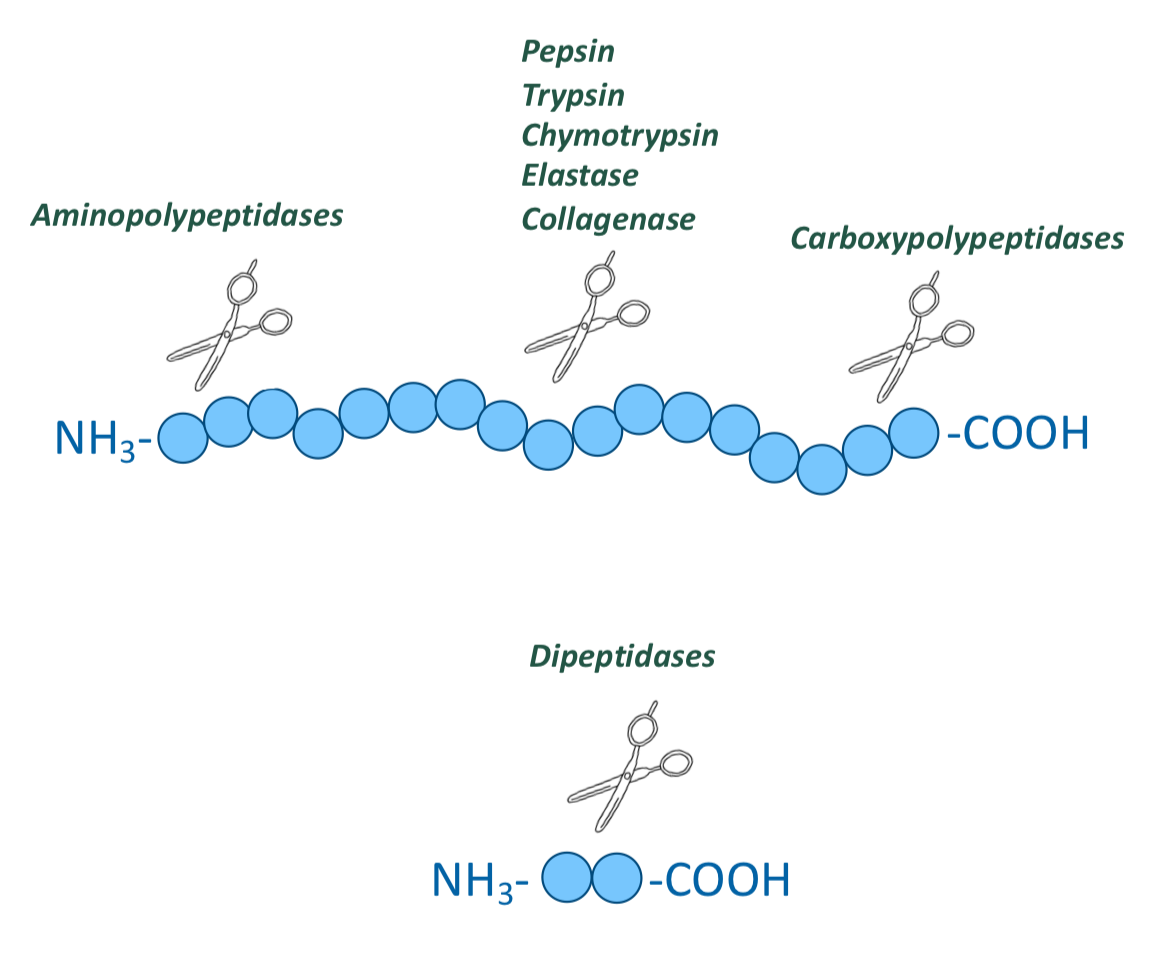
What are the peptidases in the enterocytes that line the small intestinal villi?
Aminopolypeptidases and dipeptidases
Both are exopeptidases
Hydrolyze one amino acid at a time from the N-terminal ends
They produce tripeptides, dipeptides, and amino acids
How are L-amino acids absorbed into the cell?
Via secondary active transport using Na+-AA cotransporters
There are 4 separate cotransporters for neutral, acidic, basic, and glycine/imino amino acids.
Once in the cell, the cross the basolateral membrane into the blood via facilitative diffusion
How are dipeptides and tripeptides absorbed into the cell?
Via secondary active transport using H+-dependent cotransporters
Inside the cell, the peptides are hydrolyzed to amino acids by cytosolic peptidases
They cross the basolateral membrane into the blood by facilitative diffusion
Hartnup disease - cause
Defect in the absorption of non polar amino acids
AR mutation in SLC6A19 gene
Hartnup disease - pathogenesis
Defective absorption of neutral AA (I, L, F, T, W, V, A, S, Y) from epithelial cells of intestines and kidneys
Deficient W → deficient serotonin, melatonin, and niacin
Hartnup disease - symptoms
pellagra-like symptoms (diarrhea, dementia (and other neurological symptoms), dermatitis), aminoaciduria
Infant form: photosensitivity, intermittent ataxia, tremor
Cystinuria - cause
Defect in the transport of cysteine, ornithine, lysine, and arginine (COLA)
AR mutation in SLC3A1 and SLC7A9
Cystinuria - pathogenesis
defective transport in kidney and intestine
cysteine molecules are not filtered by the kidney can be oxidized in urine to form cystine → form hexagonal white crystals, which grow into pink or yellow stones
Cystinuria - symptoms
kidney stones - may block urinary tract, aminoaciduria (but no symptoms of AA deficiency)
Summary of digestion and absorption of amino acids
Nitrogen balance
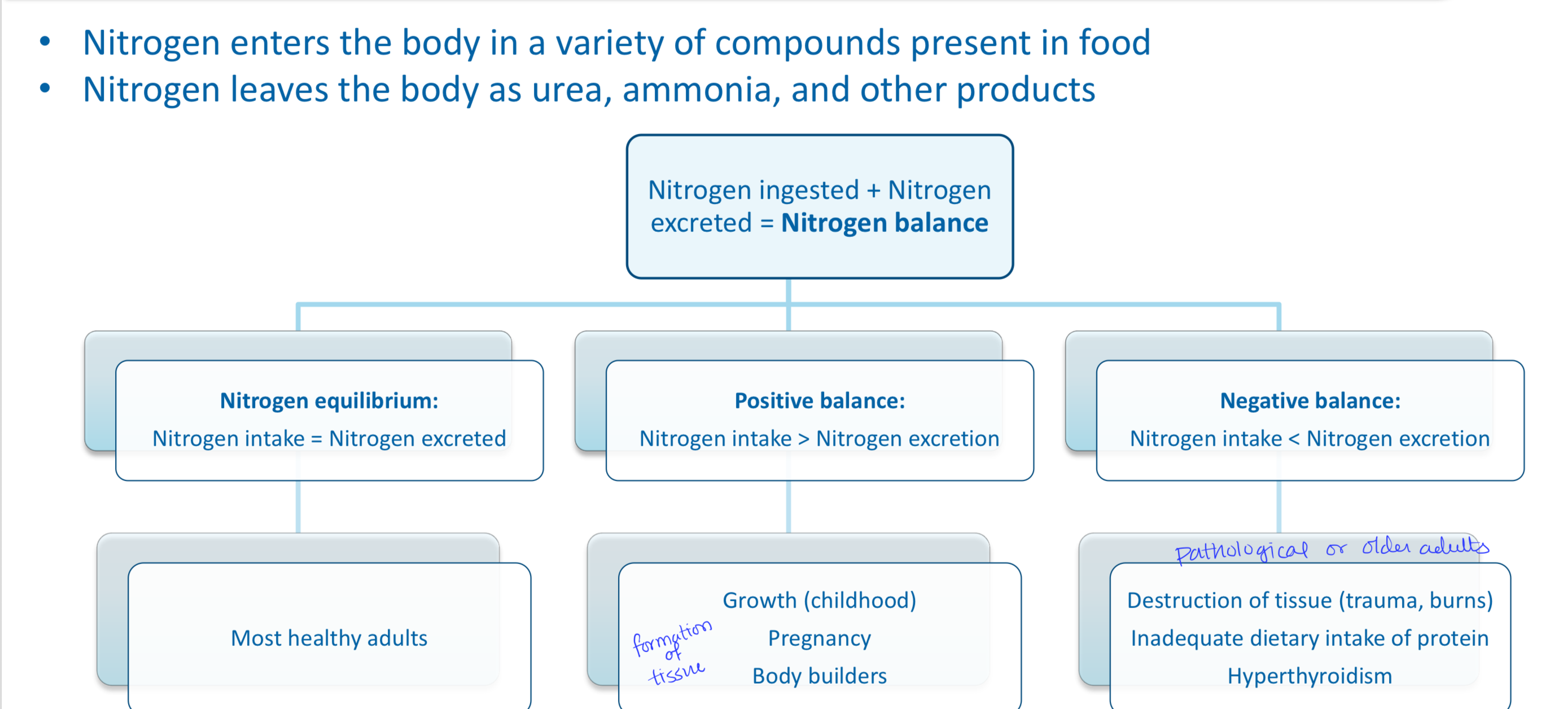
What is the amino acid pool? Explain the input and output of amino acids.
Free amino acids are present throughout the body (normally input = output)
Supplied by (input):
Exogenous (dietary) protein
Nonessential AA synthesized from simple intermediates
Degradation of body proteins
Used for (output):
Synthesis of body protein
Synthesis of nitrogen-containing small molecules
Synthesis of glucose, fatty acids, and ketones
Oxidation to CO2 and H2O
What cofactor is required for active transport into tissues?
Amino acids are transported into tissues actively using PLP (vitamin B6)
Protein turnover
Most proteins are constantly being synthesized and degraded (~300-400 g/day)

Catabolism of amino acids is important for
synthesis of many other molecules and energy
What modifications must be done to amino acids before they can be used for other pathways or energy?
AA must first undergo removal of nitrogen. The a-NH3 group prevents oxidation of AA.
Nitrogen can be incorporated into other compounds or excreted (as urea)
How are carbon skeletons of amino acids metabolized?
Various pathways:
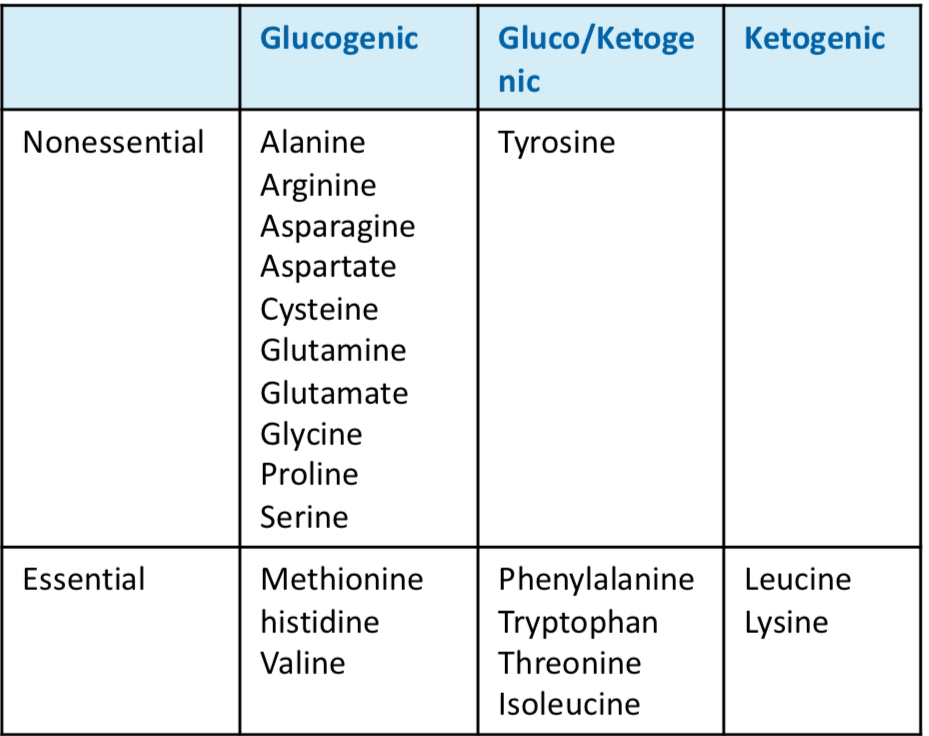
Glucogenic AA form pyruvate or intermediates of TCA cycle and can enter gluconeogenesis or energy synthesis
Ketogenic AA form acetyl-CoA, acetoacetyl CoA, or acetoacetate and can enter ketogenesis or energy synthesis
Binding of amino acids to carbon skeletons forms new AA
Removal of nitrogen from amino acids involves two steps:
Transamination and deamination
Transamination: transfer of a-amino group to a-ketoacid producing a new a-amino acid and new a-ketoacid
Deamination: oxidative deamination (requires molecular oxygen) and non-oxidative deamidation
Describe the transamination reaction of the removal of nitrogen from amino acids
reversible reaction
a-NH2 group of one a-amino acid (donor AA) is transferred to a a-ketoacid (acceptor) resulting in formation of a new a-amino acid and a new a-ketoacid
Exists for all amino acids except lysine and threonine
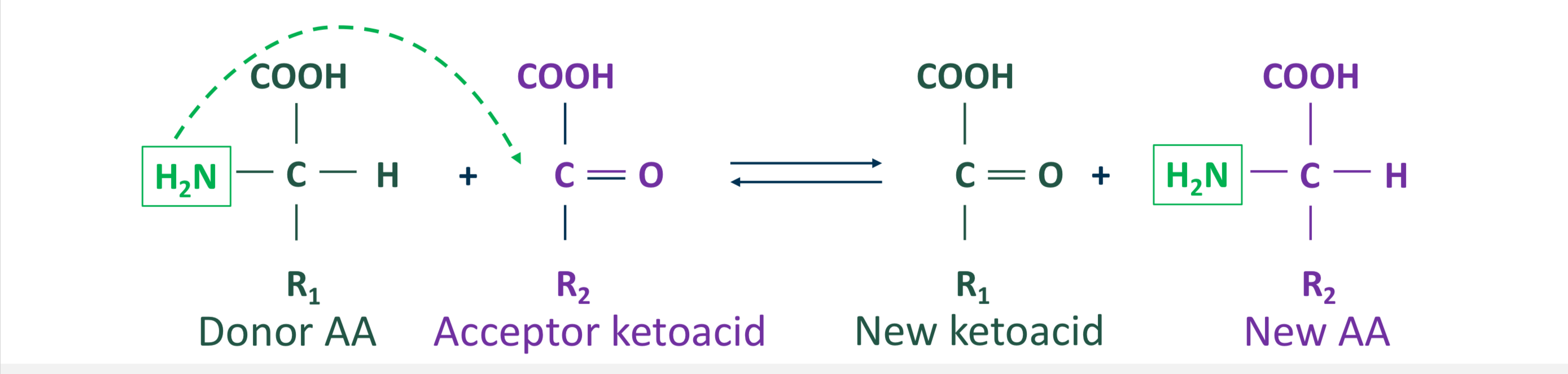
In transamination reaction, the acceptor is a ___ and the new AA is ___. Reaction is catalyzed by ___.
The acceptor is a-ketoglutarate and the new AA is glutamate. Reaction is catalyzed by transaminases (aka aminotransferases)
Where are aminotransferases located? What is the required coenzyme for this reaction?
Present in almost all mammalian tissues, in both cytosol and mitochondria. It is named after amino acid donor. Aminotransferases require PLP (B6) and there is no free ammonia formation from this reaction.
What are the two transaminases of clinical importance?
Alanine transaminase (ALT, glutamate-pyruvate transaminase) and aspartate transaminase (AST, glutamate-oxaloacetate transaminase)

What does elevated ALT or AST mean?
Small amounts detected in blood reflects damage to tissues rich in these enzymes (such as liver)
AST is also used as cardiac marker
Describe the oxidative deamination reaction of the removal of nitrogen from amino acids (GDH)
The amino groups of most amino acids are ultimately funneled to glutamate → the only AA that undergoes rapid oxidative deamination
Enzyme: glutamate dehydrogenase (GDH) or L-amino acid oxidase and D-amino acid oxidase
Glutamate dehydrogenase reaction
Reversible reaction that occurs in the mitochondria of liver and kidneys
Glutamate → a-ketogluterate
Requires NAD+
Stimulated by ADP, GDP
A-ketogluterate → glutamate
Requires NADPH
Stimulated by ATP, GTP
Releases free ammonia that enters the urea cycle
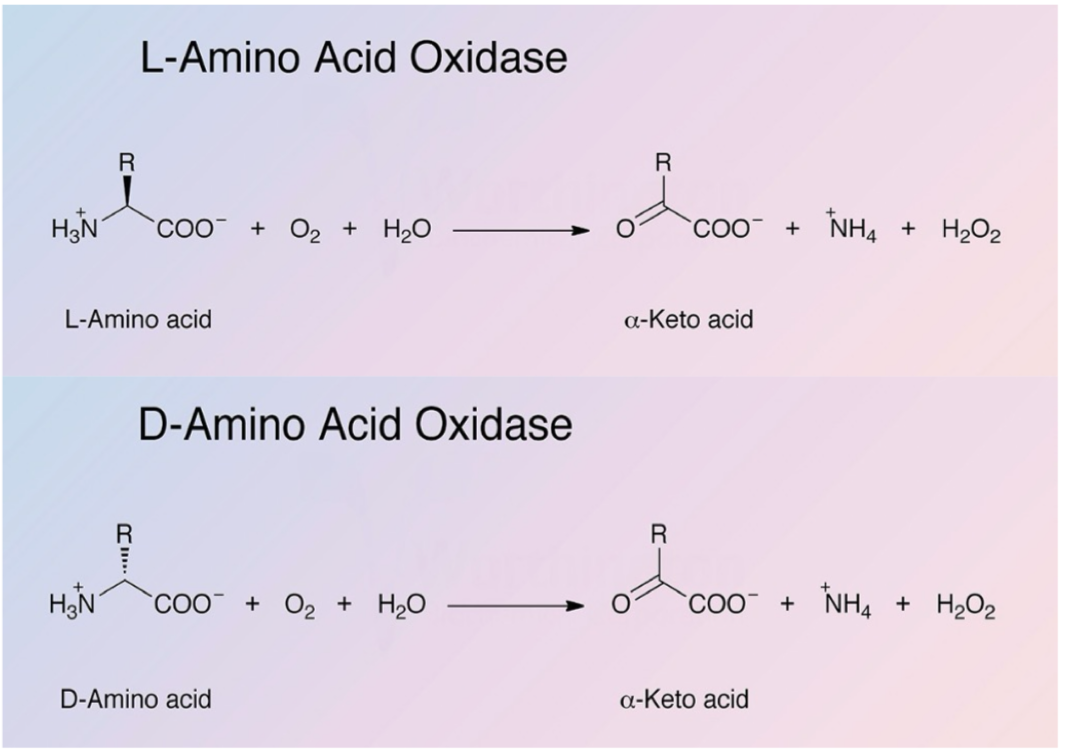
Describe the oxidative deamination reaction of the removal of nitrogen from amino acids (L/D-AA oxidase)
Enzyme: L-amino acid oxidase (requires FMN) and D-amino acid oxidase (requires FAD)
Present in peroxisomes of liver and kidneys
Produces H2O2, NH4, and keto acids
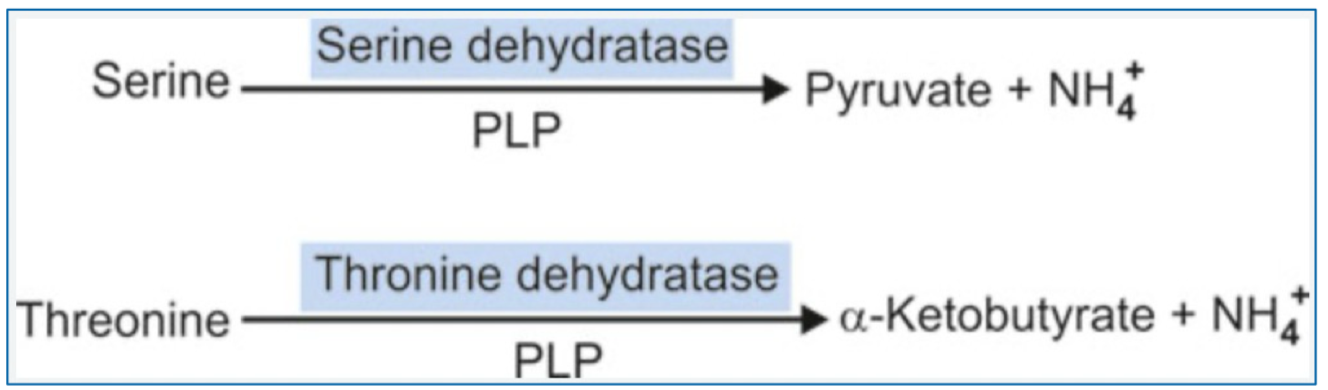
Describe the non-oxidative deamination reaction of the removal of nitrogen from amino acids. (S, hS, T)
Hydroxy amino acids deamination (AA with one or more hydroxyl groups)
Includes: serine, homoserine, threonine
Enzymes - ~amino acid~ dehydratases
Requires PLP as cofactor
Releases NH4
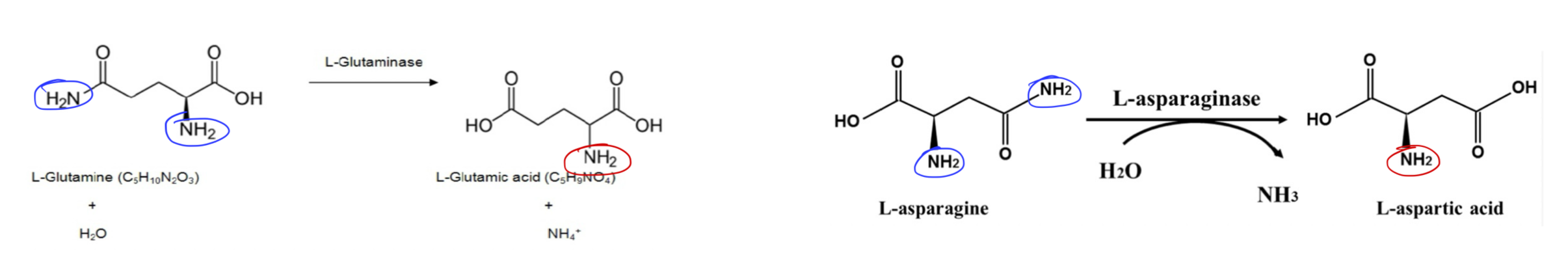
Describe the non-oxidative deamination reaction of the removal of nitrogen from amino acids. (N, Q)
Hydrolytic deamination (AA with amide groups)
Includes: asparagine and glutamine
Enzymes: hydrolases:
Glutaminase: converts Q → E + NH3 (in the liver, enters urea cycle)
Asparaginase: converts N → D + NH3
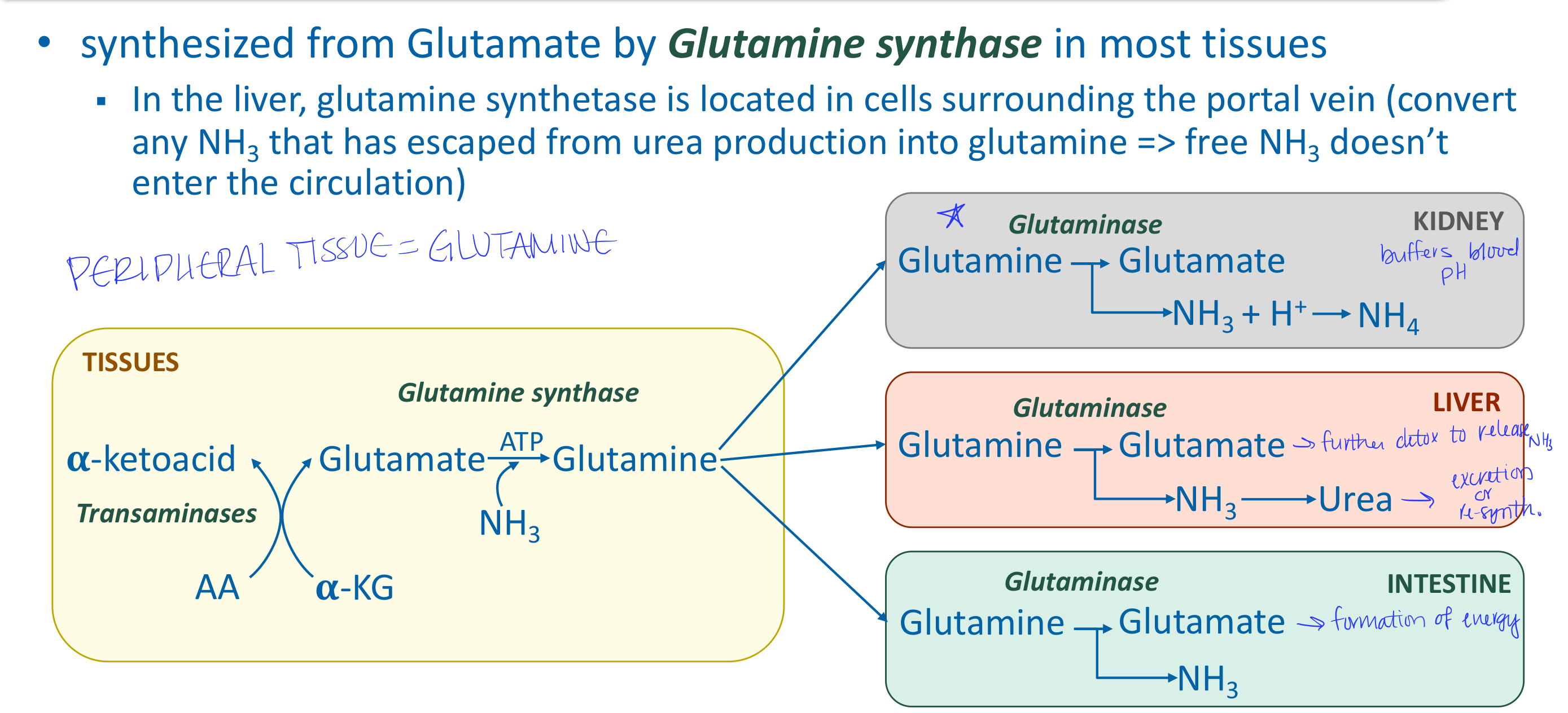
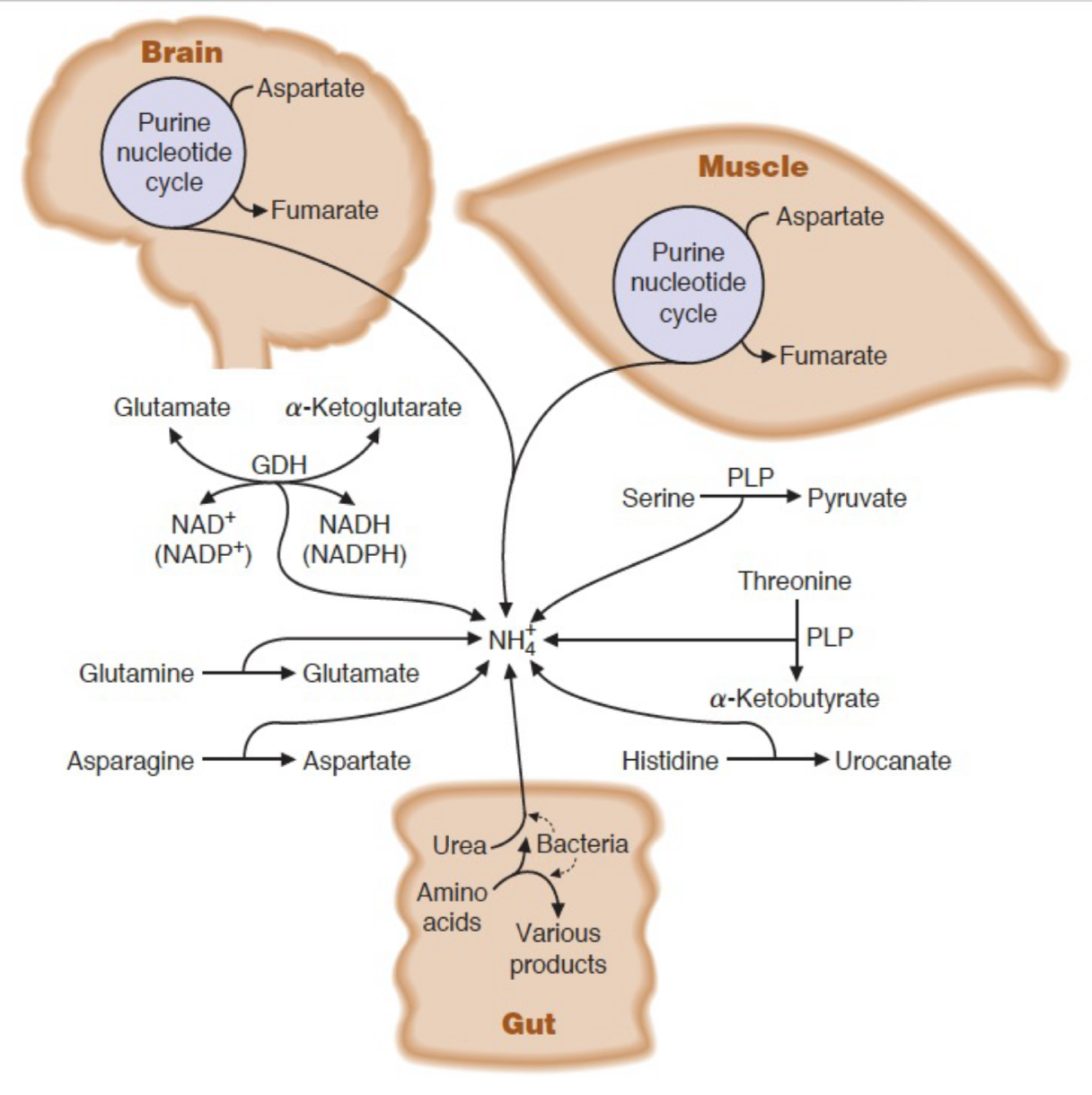
Describe the release of ammonia in most tissues
AA transamination produces glutamate → glutamine (tranport form of ammonia) → to the kidneys (buffering), liver (urea formation), intestine (used as fuel)
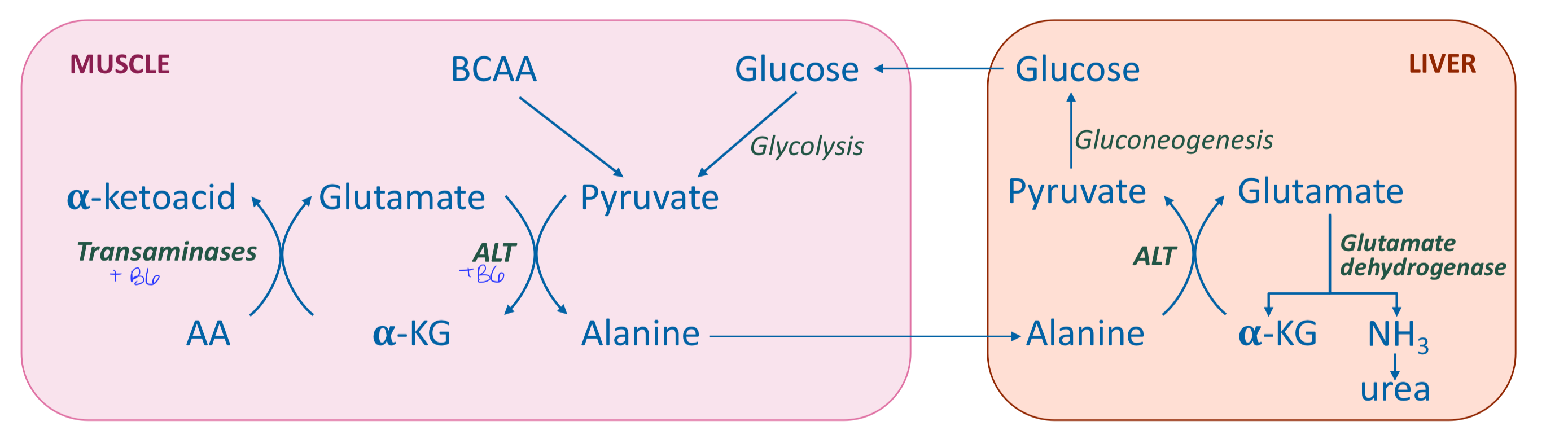
Describe the release of ammonia in muscle
AA transamination produces alanine (transport form of ammonia to liver) → produces glutamine (as other tissues) → can utilize branched chain amino acids
Describe the release of ammonia in intestines
Predominantly use glutamine and asparagine for energy → release ammonia directly to portal system
What are the major carriers of nitrogen in the blood?
Alanine and glutamine
What are the misc. sources of ammonia for the urea cycle?
dehydratase reactions of serine and threonine
deamination of histidine
purine nucleotide degradation in muscle and brain
intestinal bacteria
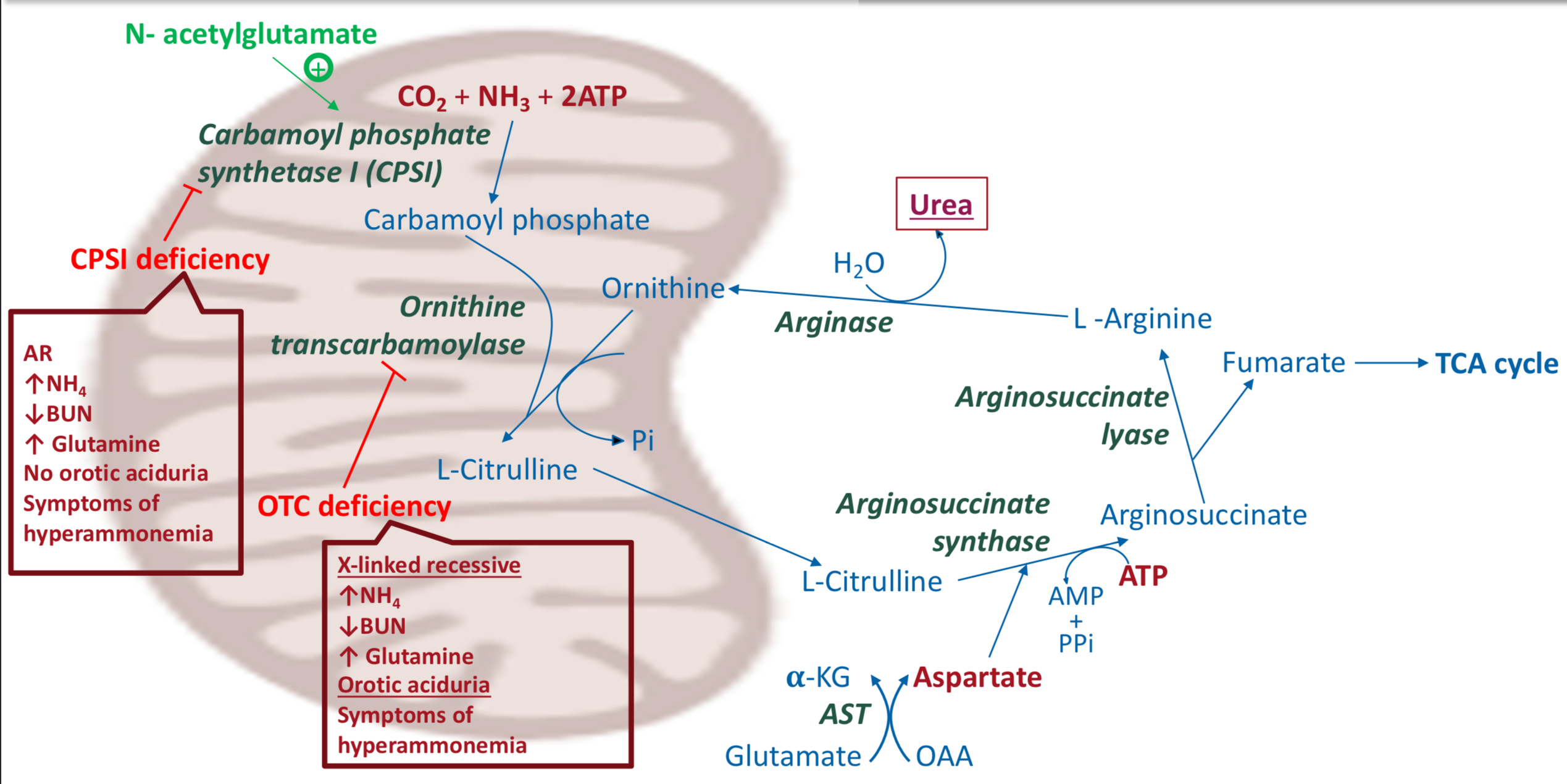
What is urea?
The major form of excreted nitrogen in humans - water-soluble and nontoxic.
Where is urea produced?
In the liver, both mitochondria and cytosol, by the urea cycle
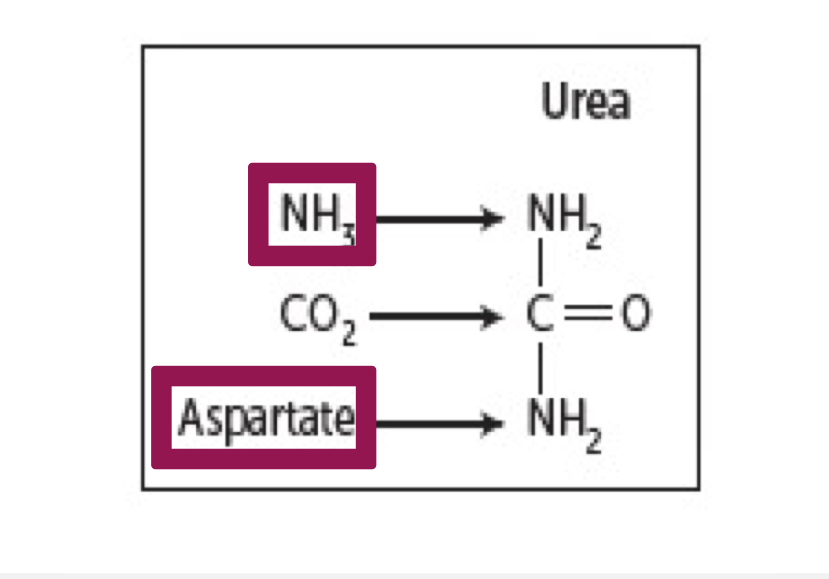
What are the donors of ammonia for urea?
1 nitrogen is from free NH3 (released from glutamate by glutamate dehydrogenase)
2 nitrogen are from aspartate (formed from transamination of glutamate by AST)
Glutamate is the final carrier of NH3
The C and O of urea are from CO2 (as HCO3-)
Symptoms of hyperammonemia (in general)
Tremors, slurred speech, somnolence, vomiting, cerebral edema, blurred vision, usually associated with decreased blood urea levels
Causes of hyperammonemia
Acquired: liver disease (acute, chronic)
Hereditary: genetic defects of urea cycle
Relationship between hyperammonemia and BUN levels
Usually, hyperammonemia is associated with decreased BUN levels
In renal failure → increased blood urea b/c it is unable to exert → diffuses into gut → urease → increased ammonia → secondary hyperammonemia
What can be used to treat/limit secondary hyperammonemia as a result of renal failure?
Oral neomycin
In renal failure → increased blood urea b/c it is unable to exert → diffuses into gut → urease → increased ammonia → secondary hyperammonemia
Causes of ammonia toxicity (3 possible results)
Increased NH3 concentration:
Enhances utilization of a-KG depressing TCA cycle
Enhances glutamine formation from glutamate
Decreased formation of GABA
Cerebral edema d/t glutamine-induced osmotic shifts
Increased outflow of glutamine from brain cells → entry of tryptophan into brain cells → increased serotonin
NH3 is basic → alkalization of intracellular compartment
Ornithine transcarbamoylase deficiency - causes
X-linked recessive defect in OTC
Ornithine transcarbamoylase deficiency - symptoms
Usually observed first few days of life
cerebral edema, lethargy, convulsions, coma
Ornithine transcarbamoylase deficiency - lab presentation
Increased NH3
Decreased BUN
increased blood glutamine
Orotic acuduria: increased blood orotic acid level because carbamoyl phosphate is accumulated → travels to cytosol → enters process of synthesis of pyrimidine nucleotides → intermediate orotic acid is accumulated
Carbamoyl phosphate synthetase deficiency - cause
Autosomal recessive defect in CPSI
Carbamoyl phosphate synthetase deficiency - symptoms
Same as ornithine transcarbamoylase deficiency
Usually observed first few days of life
cerebral edema, lethargy, convulsions, coma
Carbamoyl phosphate synthetase deficiency - lab presentation
Increased NH3
Decreased BUN
Increased blood glutamine
NO OROTIC ACIDURIA
Urea cycle enzyme deficiencies - citrullinemia
Defect in arginosuccinate synthase
Citrullinuria
Urea cycle enzyme deficiencies - arginosuccinic aciduria
Defect in arginosuccinate lyase
Arginosuccinic acid (arginosuccinate) is found in blood, CSF, and urine
Urea cycle enzyme deficiencies - hyperargininemia
Defect in arginase
Tx: diet without R
Treatment of hyperammonemia may include
Decreased protein intake
Removal of ammonia
Sodium benzoate + glycine → hippuric acid → excretion
Phenylacetate + glutamine → phenylacetyl glutamine → excretionS
Summary of amino acid metabolism
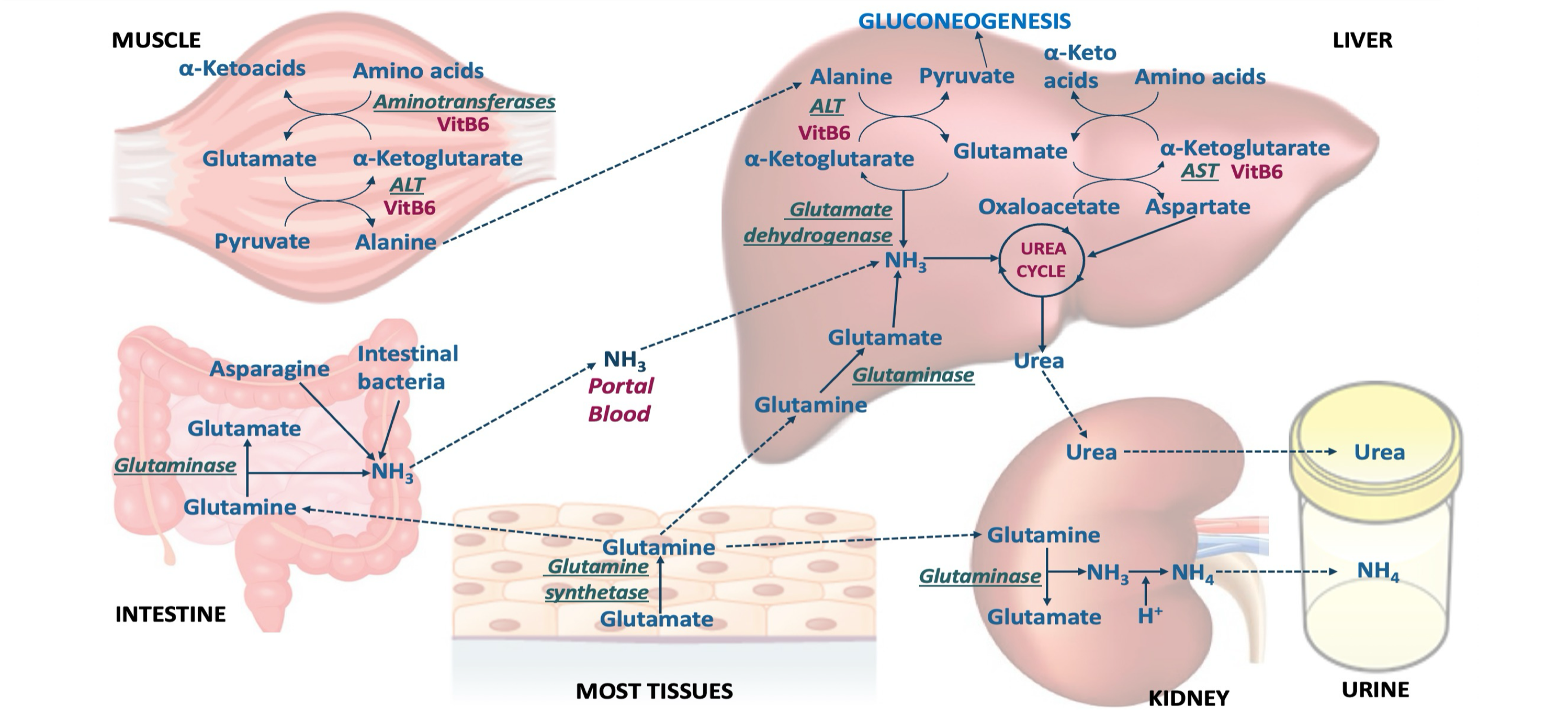
Summary of urea cycle