Heme P1
1/186
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
187 Terms
Hemolytic Anemia
Results when the rate of RBC destruction exceeds the rate of RBC production by BM
Acute hemolytic anemia
Rapid onset, isolated or episodic. Example such as hemolytic transfusion reaction resulting from a single incident of incompatible transfusion blood units
Chronic hemolytic anemia
Occurs over time. Multiple hemolytic crises or episodes. Anemia may not be evident if BM can compensate. Example includes G6PD def causing RBC lifespan chronically shortened. A complication of hgbS due to shortened RBC survival as spleen removes sickle cells.
Inherited hemolytic anemia
Passed to offspring by mutated genes from patients. Most often lead to chronic hemolysis. Example includes SCA, caused by mutated hgb gene
Acquired hemolytic anemia
Develops in previously haematologically normal individuals. An agent or condition is acquired by the individual that ultimately leads to the lysis of their RBCs. Most often lead to acute hemolysis. Exmaple includes malarial infection.
Intrinsic hemolytic anemia
Defect is present within the RBC itself. If transfused cells with defect into another individual, they would still have shorted lifespan in new host as the defect is specific to cell pop. Example includes conditions where RBC have abnormal membrane, globin or enzyme defects (Thalassemia, hereditary spherocytosis)
Extrinsic hemolytic anemia
Conditions arising from outside of RBC. Affected by factors within the patients body/circulatory system. If the patient is transfused with healthy RBC they would be affected by the destruction. Example includes anemia caused by person’s antibody in their plasma against RBC Ags.
Intravascular hemolysis
Site of hemolysis that occurs within the bloodstream. Can occur normally in the body (10%). Excessive hemolysis leads to decreased free haptoglobin due to increased hgb. Unbound hgb rapidly oxidizes to methemoglobin, which binds to hemopexin and captured by hepatocyte → iron released from metheme and protoporphyrin ring converted to conjugated bilirubin.
Free metheme and hgb can enter urinary filtrate is complexes are overwhelmed and appear as urine sediment (hemosiderin) can be detected w/ Prussian blue stain
Extravascular hemolysis
Site of hemolysis that occurs outside of the bloodstream (i.e. spleen, liver) from RBCs being engulfed by macrophages. Normally occuring in the spleen to recycle old RBC (~120 days). Excessive hemolysis caused by pathological process that lead to over expression of aged cell marker. Causes increased urobilinogen in blood and urine. Can also lead to spherocytes from partially ingested RBCs that reseals.
Normal extravascular hemolysis
When old RBCs that loses deformity is filtered out by spleen and phagocytized by macrophages. Hgb broken down into heme and globin molecules → globin durther degraded to aa. Iron released from heme and returns to plasma via transferring and recycled into cell.
Protoporhyrin converted to unconjugated bilirubin and released into blood → binds to albumin and transported to liver → enters hepatocytes and converted to conjugated bilirubin → leaves liver in bile → Bacteria in large intestine converts it to urobilinogen and excreted as stool
Normal Intravascular hemolysis
Few RBCs lyse in circulation, forms fragments → releases hgb into blood which can induce damage to cells. Haptoglobin binds hgb, and the complex binds to macrophages and degrades hgb to heme, releasing iron. Released protoporphyrin ring converted to unconjugated bilirubin. Haptoglobin then degraded in the macrophages
Intravascular hemolysis Lab results
Includes general hemolytic anemia findings: Decreased hgb and HCT, RBC count, increased total bilirubin, unconjugated bilirubin and urobilinogen.
Additional findings: Drastic decreased haptoglobin, increased plasma and urine hgb/metheme. Presence of hemosiderin in urine, fragments in PBF.
Extravascular hemolysis lab findings
Includes general hemolytic anemia findings: Decreased hgb and HCT, RBC count, increased total bilirubin, unconjugated bilirubin and urobilinogen.
Additional findings: spherocytes on the peripheral blood smear
Differential diagnosis of HA vs other cause of Bilirubinemia and reticulocytosis
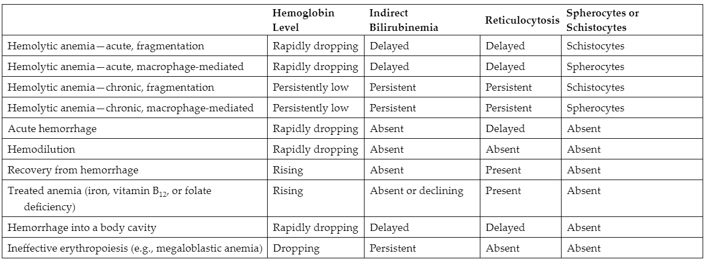
Erythropoiesis lab findings
Assuming BM is healthy, increased erythropoiesis is often first sign of hemolytic anemia. Variable WBC/PLT, decreased RBC, Hgb, HCT, increased MCV, RDW, retic and NRBC
Hemolytic anemia morphologic abnormalities
Spherocytes → hereditary spherocytosis or burn
Elliptocytes → hereditary elliptocytosis
Burr cells → pyruvate kinase def, uremia
Schistocytes → microangiopathic hemolytic anemia
Agglutination → cold agglutinins, immunohemolytic disease
RBC membrane
Shape of RBC is due to vertical and horizontal interaction of proteins including: Ankyrin and actin complex (vertical), protein 42, α & β spectrin (horizontal), G3PD. Provides deformability, elasticity and stability to cell.
Hereditary spherocytosis
Hemolytic anemia characterized by mutation (hereditary) that alter membrane structure. 75% autosomal dominant, 25% non-dominant. Mutation in protein that disrupts vertical membrane linkages btwn transmembrane and underlying cytoskeletal protein, causes loss of membrane and lower SA to volume ratio. Characterized by spherocytes and polychromasia (HJB, pap bodies and target if post-splenectomy). Features splenomegaly, jaundice, anemia. Causes increased MCHC, RDW, retic count, decreased hgb.
Osmotic fragility test
Blood added to series of tubes with gradient of NaCl sol. As RBCs are subjected to increasing hypotonic sol, they swell as water enter cell to dilute cell contents and lyses cell when volume is too large. Spherocytes will lyse in less hypotonic sol compared to normal RBC due to lower SA:vol ratio. Not specific to hereditary spherocytosis as increased fragility is demonstrated in other conditions that contain spherocytes.
Direct Antiglobulin Test (DAT)
Used to distinguish hereditary spherocytosis from an immune related HA that contains spherocytes. Detects IgG or complement on the RBCs in vivo. Immune disorder with spherocytes are usually DAT positive in testing.
Eosin-5-maleimide binding (EMA) test
Uses fluorescent dye that binds transmembrane proteins. Can also distinguish immune hemolysis from hereditary spherocytosis. Using flow cytometry, specimens with hereditary spherocytosis shows lower mean fluorescence compared with patients with immune mediated hemolysis
SDS-PAGE test
Electrophoretic separation of proteins to identify membrane protein def. Can quantify proteins present with densitometry.
Hereditary Elliptocytosis
Hemolytic anemia characterized by mutation in protein that disrupts the horizontal linkages in cytoskeleton; loss of mechanical stability of membrane. Autosomal dominant. Displays few to 100% elliptocytes on PBF with schistocytes in severe cases. Can also see retic and decreased haptoglobin levels if mod to severe anemia present. 90% of cases asymptomatic.
Hereditary pyropoikilocytosis
Rare subtype of hereditary elliptocytosis. Mutation in spectrin that disrupts horizontal linkages in cytoskeleton, causing severe RBC fragmentation. Displays elliptocytes, schistocytes, microspherocytes. decreased EMA fluorescence to differentiate from thermal injury/burn
Overhydrated hereditary stomatocytosis
Mutation in protein (Rh-associated glycoprotein) that causes increased membrane permeability to Na and K; high intracellular Na causes influx of water, leading to increased cell volume (high MCV) and lower cytoplasmic viscosity (lower MCHC). Displays stomatocytes and macrocytes.
Dehydrated hereditary stomatocytosis
Mutation in protein that causes increased membrane permeability to K. Low intracellular K causes loss of water from cell, leading to lower cell volume (low MCV) and higher cytoplasmic viscosity (high MCHC). Displays target cells, burr cells, stomatocytes, “puddled” hgb at periphery.
Rh deficiency syndrome
Rare hereditary condition in which expression of Rh membrane proteins on RBCs is absent. Mild to mod HA. Stomatocytes and occasional spherocytes seen on PBS. Treated by splenectomy.
Acquired stomatocytosis
Associated with acute alcoholism and certain medications. May see drying artifact on Wright stained PBF.
Glucose-6-Phosphate Dehydrogenase (G6PD)
Needed to protect hgb, protein and lipids from oxidative denaturation. Catalyzes the first step in a series of reaction that detoxify the hydrogen peroxide that is formed from oxygen radicals. Only means of generating NADPH in the monophosphate shunt which is required for detoxification.
G6PD deficiency
Deficient RBCs cannot make enough NADPH to effectively detoxify H2O2 during oxidative stress, causing oxidative damage of hgb and membrane, converting hgb to metheme which creates Heinz bodies (insoluble denatured hgb) which causes irreversible damage. Heinz bodies are removed mostly by intravascular hemolysis. Shows X linked inheritance pattern. Treated by discontinuing oxidative agent such as drug and treating underlying infection or transfusion of RBCs to treat anemia.
G6PD deficiency manifestation
Individual with mild to mod def can be clinically normal until they come in contact with certain drugs, infections or food, and hemolysis can begin abruptly or gradually. Also associated with neonatal hyperbilirubinemia. WBC count is mod. elevated, PLT count variable, hgb decreased, retic count increased. Serum haptoglobin severely decreased with hemoglobinemia and hemoglobinuria present. PBF shows bite and blister cells, increased polychromasia, spherocytes and schistocytes. Heinz bodies seen in supravital stain.
Pyruvate kinase (PK)
Key enzyme of the glycolytic pathway. Catalyzes the conversion of phosphoenolpyruvate to pyruvate forming ATP.
PK deficiency
Causes premature destruction of RBCs due to depletion of ATP resulting in lack of membrane integrity. Presents as anemia, jaundice, splenomegaly, gallstones.
PK deficiency lab findings
Normal of slightly increased WBC and PLT, variable hgb, increased retic. Decreased serum haptoglobin, increased serum indirect bilirubin and urine urobilinogen. PBF shows polychromasia and burr cells alongside anisocytosis and poik. Confirmed by PL activity assay (decreased) and genetic testing. Can be treated increasing 2,3 BPG level → promotes greater release of O2 at tissue to tolerate lower hgb levels to prevent tissue hypoxia.
Paroxysmal nocturnal hemoglobinuria (PNH)
Rare chronic intravascular hemolytic anemia. Mutation in a clonal hematopoietic stem cell that results in circulating blood cells that lack surface marker CD55 and 59, which are complement inhibiting proteins. Cell cannot prevent activation of complement without them, causing spontaneous intravascular hemolysis. Symptoms include dark first morning urine, muscle dystonia, hepatic vein thrombosis.
PNH lab findings
Decreased serum haptoglobin, increased plasma hgb, serum indirect bilirubin. Hemoglobinuria and hemosiderinuria present. Increased retic, and MCV, pancytopenia, eventual IDA due to urinary loss of iron. Confirmed by Ham’s test, sugar water test, and flow cytometry. Treated by Eculizumab → monoclonal Ab against complement C5 to inhibit complement (does not address BM failure and complications)
Immune hemolytic anemias
Conditions where RBC survival is shortened due to Ab mediated mechanisms. Some Abs can activate complement to attach to RBC membrane, leading to premature removal by macrophages (extravascular), complement mediated hemolysis (intravascular) or a combination. Classified as autoimmune, alloimmune, and drug induced. IgM mediated can result in both extra and intravascular hemolysis, IgG is predominantly extravascular hemolysis.
IgM mediated hemolysis
If present in low density on RBC membrane due to small immune response, complement activation starts, but cannot reach full activation. Causes RBCs to be destroyed by extravascular hemolysis. If large immune response occurs and high density is present on RBC, complement proceeds fully and intravascular hemolysis occurs.
IgG mediated hemolysis
Occurs with or without complement through extravascular means. RBCs will be removed by macrophages in spleen when attached with Ab as they have IgG receptors. If high density is present, complement can partially activate where hepatic macrophages will detect complement protein and clear the RBCs. Cells are sometimes partially phagocytized, allowing membrane to reseal to form spherocytes (characteristic), with normal or micro size.
Immune HA lab findings
WBC and PLT possible increased, decreased hgb, increased retic. Serium haptoglobin decreased, serum unconjugated bilirubin and plasma hemoglobin (intravascular) increased. Hemoglobinuria and hemosiderinuria seen. Polychromasia, spherocytes, NRBCs, fragments observed with possible agglutination and leukocytosis/thrombocytosis. Confirmed by DAT positive
Autoimmune hemolytic anemia (AIHA)
Premature RBC destruction and anemia caused by autoandibodies that bind to the RBC surface with or without complement activation. Divided into WAIHA, cold agglutinin disease, paroxysmal cold hemoglobinura, mixed AIHA
Warm autoimmune hemolytic anemia (WAIHA)
Most common AIHA. Classified as idiopathic (unknown cause) or secondary (patient with other conditions such as leukemia). Most cause due to IgG autoantibodies and extravascular hemolysis with optimum reactivity of 37C. can be detected through DAT (IgG or IgG + complement). Lab findings include polychromasa and spherocytes. Treated by glucocorticosteroid, transfusion (unless broad autoantibody), splenectomy (50% effective), Rituximab (new, not well known).
Cold Agglutinin disease
IgM autoantibodies bind RBCs when patient exposed to cold temp, and activates complement. IgM will dissociate when temp returns, but complement will stay attached. Hemolysis occurs through hepatic macrophage, but some intravascular is possible from full complement. Complement sensitization detected by DAT. Lab findings include agglutination and hemoglobinuria. Usually self limiting and require no treatment. Transfusion, supportive care, Rituximab (effective 1 year), plasmapheresis (temp reduction of auto IgM) if severe.
Paroxysmal cold hemoglobinuria (PCH)
Cold reactive hemolytic anemia by IgG. Usually caysed by Anti-P auto-Ab that binds P Ag on RBC in cold temp and partially activates complement. Complement is then fully activated at warm temp and hemolyzed via intravascular. Common in young children after resp infection. Usually self limiting, though severe. Transfusion can be given until resolved.
Mixed type autoimmune HA
Patient develops both IgG auto-Ab (reactive at 37C like WAIHA) and IgM auto-Ab (reactive at 0-10C). Results in combination of extra and intravascular hemolysis. Usually involves chronic disease course with episodes of severe anemia.
Hemolytic transfusion reaction
Type of alloimmune HA. Immune mediated destruction of donor cells by an Ab in recipient. Can be acute and delayed onset.
Acute Hemolytic Transfudion reaction (AHTR)
occurs within minute to hours of initial transfusion. Common cause is accidental transfusion of ABO incompatible donor where recipient’s preformed IgM Ab to donor cell causes complement mediated intra hemolysis. Lab findings include hemoglobinuria and hemoglobinemia, decreased hgb and serum haptoglobin, increased serum indirect bilirubin, positive DAT. Patient should be monitored for DIC with coag tests.
Delayed hemolytic transfusion reactions (DHTR)
May occur days or weeks after transfusion. Usually patient has been exposed to Ag in the past and the Abs generated were below serological detectable levels initially. Subsequent exposure to the same Ag will increase alloantibody to levels where they can bind to transfused RBCs and cause extravascular hemolysis. Signs include inadequate post transfusion increase in hgb, positive DAT in IgG/complement, morphologic evidence of hemolysis, increase in serum unconjugated bilirubin.
Hemolytic disease of the fetus and newborn (HDFN)
IgG alloantibody produced by mom crosses the placenta into fetal circulation and binds to fetal cells. Sensitized fetal cells are cleared by macrophage in fetal spleen and anemia develops. BM and fetal organs will compensate and release NRBC into circulation. Includes Rh and ABO form.
Rh HDFN
Rh- mom has preformed anti Rh Ab (IgG). Mom can be exposed through previous pregnancy with Rh positive baby or through prev blood transfusion with Rh+ blood. anti-Rh alloantibody crosses placenta in subsequent pregnancy, and if fetus is Rh+, the sensitized cells are cleared by fetal spleen. Findings include decreased Hgb, increased retic and serum unconjugated bilirubin with many NRBC and polychromasia in PBF.
ABO HDFN
Seen in some type A or B infant born to type O moms who produce IgG anti-A and B Abs that crosses the placenta. Milder than Rh HDFN as A and B Ags are poorly developed in newborns. Usually asymptomatic, may have mild hyperbilirubinemia, DAT may be weakly positive or negative. PBS may contain polychromasia and spherocytes.
Drug induced hemolytic anemia (DIIHA)
Suspected when there is a decrease in hgb after administration of a drug with clinical and biochemical evidence of intra/extra hemolysis and DAT positive. Can be caused by antimicrobials, anti-inflammatories, anti-neoplastic medications. Abs implicated can be drug dependent (either react only with drug treated cells or only react in presence of drug) or drug independent
Extrinsic non-immune hemolytic anemia
Conditions that causes physical or mechanical injury to the RBCs. Can be caused by abnormalities in the microvasculature of heart and large vessels, infectious agents, chemicals, drugs, venoms, extensive burns.
Microangiopathic hemolytic anemia (MAHA)
Characterized by RBC fragmentation and thrombocytopenia. Occurs intravascularly by mechanical shearing of RBC membranes as they pass partially blocked microthrombi or damaged endothelium. Contains 4 subtypes: TTP, HUS, HELLP and DIC
MAHA lab findings
Includes classic HA evidence like decreased hgb and serum haptoglobin and increased retic, serum unconjugated bilirubin and urobilinogen. Also includes fragments and thrombocytopenia.
Thrombotic thrombocytopenic purpura (TTP)
Characterized by appearance of MA, severe thrombocytopenia and marked elevation in serum LDH. Caused by deficiency of ADAMTS13 resulting in ultra long VWF multimers that bind and activate PLT causing severe thrombocytopenia, ischemia in brain, HA due to RBC rupture. Divided into 3 types: idiopathic (auto-Abs inhibit ADAMTS13), secondary (acquired condition suppress ADAMTS13 synthesis), and inherited (mutation of ADAMTS13 gene)
TTP lab findings
Severe thrombocytopenia, marked decreased hgb, schistocytes, polychromasia and NRBC, increased wbc (immature granulocytes may be seen). In biochemistry, marked increased serum LDH, increased serum unconjugated bilirubin, decreased serum haptoglobin. Coag testing shows marked decreased ADAMTS13, normal PT/PTT (differentiates from DIC)
TTP treatment
Idiopathic: treated with plasma exchange, corticosteroids, Rituximab
Secondary: Treat primary disease first and support patient
Inherited: treated with fresh frozen plasma
ADAMTS13
protease thatregulates the size of VWF by cleaving ultra long multimers. This prevents VWF multimers from excessively binding and activating PLTs
Hemolytic uremic syndrome (HUS)
Characterized by HA, thrombocytopenia and acute renal failure (dmg to endothrlial cells in glomerular microvasculature). Caused by bacteria that produce Shiga toxin (E. coli most common). Onset of illness is acute gastroenteritis.
Shiga toxin in HUS
Absorbed from intestine into plasma. Toxin has an affinity for endothelial cells particularly in glomerulus. Toxin transported to endothelial cell where it inhibits protein synthesis and cause endothelial injury. Secretion of cytokines and the toxin induces prothrombotic changes where expression of TF and increased secretion of ultra long VWF multimers. Cell damage causes narrowing of small blood vessels, which is made worse by activation of PLT and formation of fibrin thrombi.
HUS lab findings
Decreased hgb, fragments, polychromasia with mild to moderate thrombocytopenia. Normal PT/PTT (diff from DIC). Marked increased serum LDH, increased serum unconjugated bilirubin, decreased serum haptoglobin, increased serum creatinine with proteinuria and hematuria.
Hemolysis, Elevated liver enzymes, low platelet count syndrome (HELLP)
Rare but severe complication of pregnancy. If a patient develops preeclampsia, abnormalities in the development of placental vasculature result in poor perfusion and hypoxia which causes certain anti-angiogenic proteins to release from placenta which inactivate placental and edothelial growth factors. This cause placenta to suffer from vascular insufficiency and results in maternal endothelial cell dysfunction, and activates PLT and promotes fibrin deposition, especially in liver.
HELLP lab findings
Includes anemia and ciochemical evidence of hemolysis. Schistocytes on the PBS, low PLT count, normal PT/PTT. Increased serum LDH and AST are major diagnostic criteria
Disseminated intravascular coagulation (DIC)
Characterized by systematic activation of the hemostatic system resulting in fibrin thrombi formation throughout microvasculature. Results in organ damage and eventually bleeding due to consumption of PLT and coag factors. Can result as complication from burn, venomous bites, chronic inflammation, cancer or pregnancy.
DIC lab findings
Similar lab findings to other MAHAs including increased serum/urine urobilinogen and decreased haptoglobin, hgb and increased retic. Thrombocytopenia, fragments and possible polychromasia. PT/PTT prolonged with decreased fibrinogen and increased D-dimer
Traumatic cardiac hemolytic anemia
Prosthetic cardiac valves can cause hemolysis because of turbulent blood flow going through and around implanted devices. Usually mild and compensated by BM. Fragments can be found with increased retic and other HA findings. Normal PLT count differentiates from other MAHAs.
Malaria
Potentially fatal infection of the RBCs with a protozoan parasite Plasmodium. Caused by 5 different specie: P. vivax, P. ovale, P. malariae, P. falciparum and P. knowlesi. Causes anemia by direct lysis when merozoites are released from RBCs and destruction of infected and non-infected RBCs in spleen. Falciparum particularly lethal as infected RBCs adhere to endothelial cell in internal organ esp. brain, causing cerebral malaria.
Malaria lab findings
Neutropenia with monocytosis may develop. When severe, findings include metabolic acidosis, decreased serum Glu and hgb, increased serum lactate and creatinine. Vivax and ovale infects retic only, malariae infects older RBC and falciparum infects all stages of RBCs. 2 thick and thin smears are made: thick detects parasite and thin to speciate and calculate parasitemia by counting 500 to 3000 RBCs
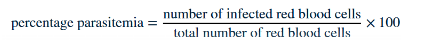
Malaria diagnosis and treatment
Rapid antigen test detects Plasmodium and must be used in conjunction with PBF due to low sensitivity. PCR can also detect and speciate. Serology only detects Abs and may not be current infection. Common treatment includes Artemisinin-based combination therapies (ACTs) and Chloroquine phosphate.
Babesiosis
Tick transmitted disease caused by intraerythrocytic protozoan parasites Babesia. Humans are incidental hosts after injection of sporozoites during blood meal of ticks. Causes mild to severe HA in 60% of infected. Lab findings includes typical HA alongside decreased haptoglobin and hgb and increased retic. Morphology similar to P. falciparum but rings are usually vacuolated with no pigment and may have formation of merozoite tetrads (Maltese cross formation)
Filariasis or elephantiasis
Parasitic disease caused by microscopic worms that only lives in lymphatic system. Does not cause HA. Standard method of diagnosis is identification of microfilariae in PBF. Blood collection should be done at night to coincide with appearance of microfilariae in circulation, which usually appears at edge of smear. Patient may exhibit absolute eosinophilia.
Clostridial sepsis
Causes massive intravascular hemolysis due to toxin that can hydrolyze RBC membrane phospholipids causing RBC to become spherical and susceptible to lysis. Sudden massive hemolysis causes dark red plasma and urine. Lab findings include very decreased HCT, spherocytes, microspherocytes and toxic change in neuts.
Drug or chemical induced HA
Causes oxidative denaturation of hgb, leading to formation of metheme and Heinz bodies. Typical HA findings are seen with bite and blister cells (Heinz bodies seen in supravital stain). Basophilic stippling may be seen if heavy metal poisoning is seen with copper and lead (usually normochromic RBCs)
Venom induced HA
Mechanism can include disruption of RBC membrane, alteration of RBC membrane resulting in complement mediated lysis, initiation of DIC or renal failure. Severity depends on amount injected and the specific mechanisms.
HA from Thermal injury
Patients present with RBC injury and acute hemolytic anemia. Lab findings include fragments, spherocytes, microspherocytes, and irregular RBC budding. Damage is usually cleared by spleen within 24 hours. Similar morph can be seen in malfunctioning blood warmer prior to transfusion.
Hemoglobin components
Globular protein with 4 heme groups and 4 polypeptide chains (globin chains). 4 globin chains comprising each hemoglobin are 2 identical pairs of different polypeptide chains
Globin chains
7 different types. Production changes with age - embryo, fetus, newborn and adult. Variations in AA sequence give rise to different types denoted by different greek letter.
Alpha-like genes
Located on chromosome 16. Includes 2 alpha and 1 zeta genes.
Beta-like genes
Located on chromosome 11. Includes 1 beta, delta, epsilon and 2 gamma genes
Hemoglobin assembly
Production of globin chains take place in erythroid precursors. Mature RBC does not have ribosomes and mitochrondria → cannot product hgb. Alpha chains are +vely charged, beta are -vely charged. Alpha has the highest affinity for beta, then gamma and delta. A alpha and non-alpha bind to 1 Heme each, then pair with another heterodimer to form tetramer of hgb.
Hgb development in first 3 months
Only 1 alpha-like (zeta) and 1 beta-like (epsilon) are active which forms Gower-one. Then alpha and gamma chain synthesis leads to production of Gower 2 and Portland, then finally fetal hemoglobin (HbF)
Hgb development 6 months after birth
Gamma chain synthesis gradually decrease and replaced by beta chain → leads to production of HgbA (2 alpha and 2 beta) which is the adult Hgb. Remaining delta gene also activated, producing low levels of delta chain which forms HgbA2 (2 delta and 2 alpha)
Hemoglobinopathies
Results from a genetic mutation in one or some of the gene that affect hgb synthesis. Includes qualitative/structural or quantitative change. Most common mutation are point mutations
Qualitative (structural) hemoglobinopathies
Hgb synthesis occurs at a normal or near normal rate, but hgb molecule has altered aa sequence within the globin chains. This change in aa sequence can alter the structure of the hgb molecule, resulting in a structural defect. It can also alter function, which is a qualitative defect
Quantitative hemoglobinopathies
Results in a reduced rate of synthesis of the globin chains (and therefore hgb), but the aa sequence are not affected.
Zygosity
Relationship btwn the number of genes mutated ad the level of severity of the genetic defect. Mutation in beta gene is more severe as there is only one other gene to compensate, whereas mutation in 1 alpha gene is not as severe.
Sickle cell disease
Homozygous for hemoglobin S or compound heterozygous (HgbS in combination with another Hgb beta chain). Caused by point mutation in the beta globin gene that results in Val replacing Glu in 6th position. Autosomal co-dominant, where homozygotes have a more severe disease than those who are heterocygotes. Higher HgbF offers some protection from disease.
Hemoglobin S
aa substitution affects the way hgb molecules interact with one another in the RBC cytosol. Val (hydrophobic) replaces Glu (hydrophilic). In normal RBC, Glu can extend outward from surface of hgb tetramer to contribute to solubility in RBC cytosol. In mutated chain, when valine is extended outward it tries to hide in hydrophobic niche instead. Oxygenated hgbS remains soluble as theres no hydrophobic niche, but in deoxygenation a hydrophobic pocket is created, and adjacent HgbS can begin to bind. Occurs in reversible and irreversible form.
Hemoglobin S pathophysiology
Crenated cell will polymerize as hydrophobic niches are created, which thickens blood and slowing blood flow → promoting hypoxic environment and causing more sickling. Reduction of pH also increases 2,3-BPG, which promotes more sickling as oxygen affinity is decreased.
Vaso-occlusive crises (VOC)
A hallmark of SCA. Triggering event that causes reversible sickle cells to change shape. This change causes a loss of cell deformability and they occlude the small capillaries and postcapillary venules. Can lead of hemorrhagic, ischemic stroke, or retinal ischemia
Splenic sequestration/Autosplenectomy
A hgbS complication. Sudden trapping of blood in the spleen, leading to rapid decrease in hgb. This repeats and causes scarring, causing abnormal tissue function which leads to gradual loss of tissue function in the spleen → auto splenectomy. Can lead further down to bacteria infection and septecemia
Acute chest syndrome
A hgbS complication. Repeated series of pulmonary infarction → decreases alveolar tensions that induce more sickle formation. This further cases vasoocclusions, decreases pulmonary blood flow, and further polymerize hgbS. Causes fever, hypoxia, chest pain, rapid decrease in hgb and PLT. Patient requires pain relief and oxygen
Aplastic episodes
A hgbS complication. SCA patients usually compensate for decreased RBC survival with increasing BM output. If BM gets suppressed temporarily by infection, HCT would decrease substantially with no retic compensation. Usually self limiting.
SCA treatment
Hydration to maintain good blood flow, avoid low oxygen environment (strenuous exercise, high altitudes etc), treatments to increase hgbF (hydrourea), prophylactic antibiotics, pain management, VOC specific treatment, transfusion, stem cell transplant and gene therapy
Sickle cell trait
A benign condition for individuals that are heterozygotes for hemoglobin S gene. Individuals usually asymptomatic unless under severe hypoxic condition. Only consistent abnormality is kidney impairment due to diminished perfusion to kidneys
Hemoglobin C disease
Caused by point mutation in the beta globin gene which replaces Glu with Lys in 6th position of aa chain. Changes the net charge of hgb molecule, results in less soluble hgb → will crystallize to form short thick crystals even in oxygenated state. Minimal disruption to RBC shape, less splenic sequestration and hemolysis with no VOC. Hb CC is mile while Hb AC is asymptomatic. Similar inheritance pattern to hgbS
Hemoglobin E disease
Caused by point mutation in beta globin gene where Lys replaces Glu in 26th position. Substitution results in overall net charge change, but due to position, does not cause polymerization of hgb molecule. Does cause reduced production to HgbE. Homozygotes have mild anemia, while hetero is asymptomatic. Similar inheritance pattern to hgbS
Hemoglobin C-Harlem
Involves double subsitution of the beta globin gene: Glu6Val and Asn73Asp. Patients are usually asymptomatic
Hemoglobin SC disease
Occurs when patient inherits HgbS from one parent and HgbC from the other → Glu6Val on one chain, Glu6Lys on the other. They do not produce any HgbA. Usually milder than SCA but can cause VOC (less frequent and damaging). Patient exhibits mod HA, splenomegaly, retinopathy and resp tract infections.