Biochem for boards Super Set
1/304
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
305 Terms
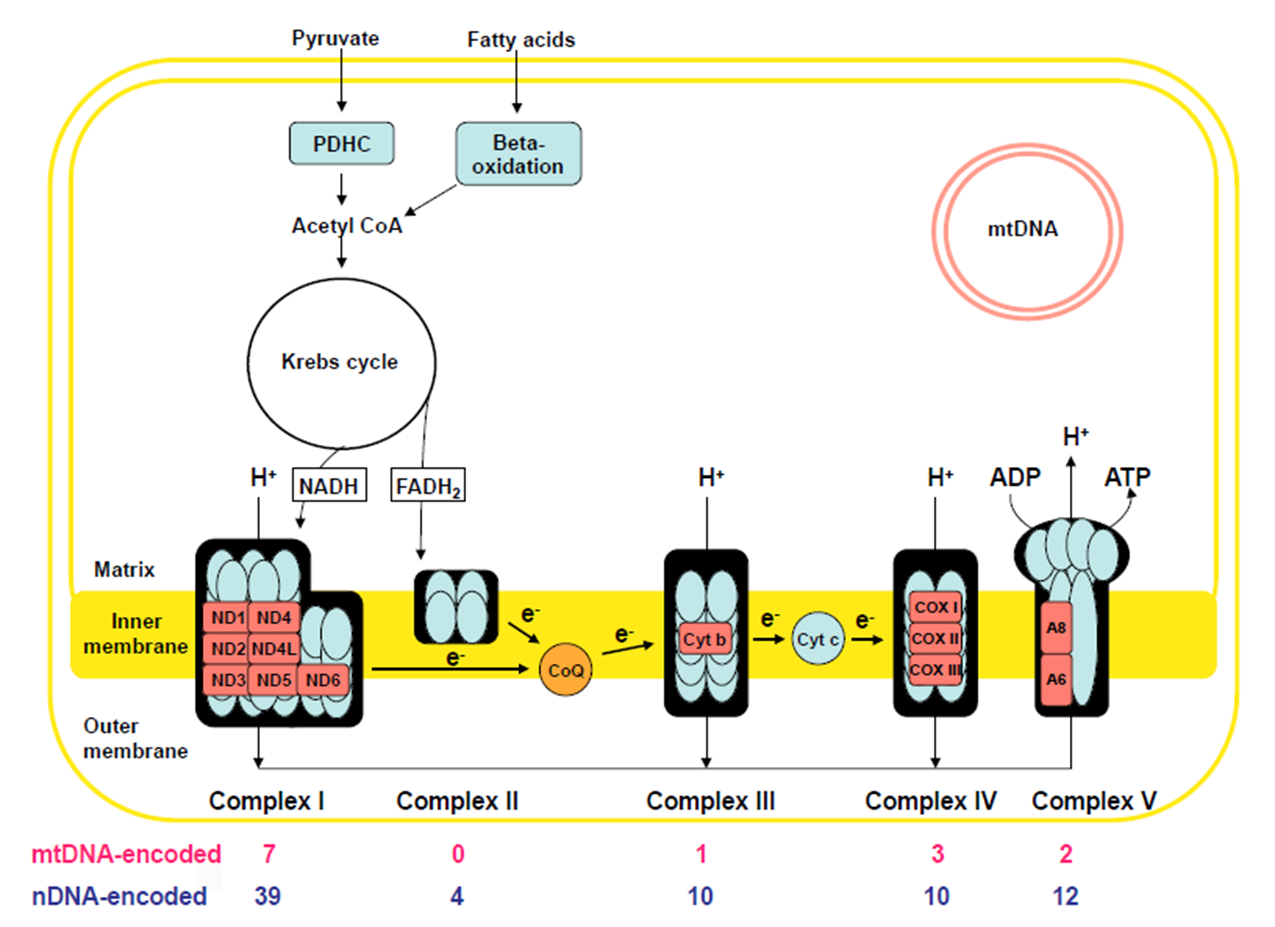
Mitochondria
Site of oxidation phosphorylation: via the electron transport chain embedded in the inner mito membrane
Produce ATP
The other biochemical processes occur in the Mito:
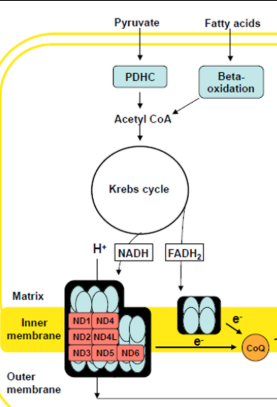
Pyruvate oxidation
Krebs Cycle
Fatty Acid Beta-Oxidation
Oxidative Phosphorylation
The electron transport chain
Inner membrane has 5 distinct protein complexes embedded: some encoded by nuclear DNA, some encoded by Mito DNA
Use NADH + FADH coming form Krebs cycle break down of Actyl-CoA
Fatty Acids (generated via F.A. B-Oxidation)
Pyruvate (generated via glycolysis)
Electron transport down the chain of complexes: creates gradient by pumping IN H+ ions
Complex V uses gradient to Generate ATP as H+ ions move OUT
Mitochondrial Disorders
Oxidative Phosphorylation/Electron Transport chain dysfunction
Two varieties:
Secondary Mitochondrial dysfunction: Non-genetic conditions
Hypoxemia (inadequate Oxygen for Oxidative Phosphorylation)
Medication: valproic acid, HIV meds
Toxins: cyanide, rotenone
Primary Mitochondrial Disease
mitochondrial DNA itself or nuclear DNA mutations
Mitochondrial Genome
The Mitochondrial Chromosome: encodes 37 genes
only 3% of Mito. proteins are encoded by mito DNA
97% are encoded by nuclear DNA and imported into mitochondria
Complex 1
46 total proteins
MtDNA encoded: 7
nuDNA: 39
Leigh Syndrome
Leukodystrophy
Complex 2
4 proteins: ALL nuDNA ENCDOED
Leigh Syndrome
Paraganglioma
Pheochromocytoma
Complex 3
11 proteins
MtDNA: 1
nuDNA: 10
Leigh syndrome
GRACILE syndrome
Complex 4
mtDNa: 3
nuDNA: 10
Leigh Syndrome
Hepatopathy
Cardioencephalomyopathy
Leukodystrophy/tubulopathy
Complex V
mtDNA: 2
nuDNA: 14
Maternal Inheritance
mtDNA mutations can only be inherited through the mother
all mito provided by the ovum
no mito contriubted by the sperm
Heteroplasmy
Mito genomes can differ between mitochondria in a given cell and % of mutant mtDNA can vary in an individual from cell-to-cell and tissue-to-tissue
Each cell has up to 1000 mitochondria, each with their own copy of the mito genome
mtDNA mutation rate is 10-20x nuclear DNA mutation rate
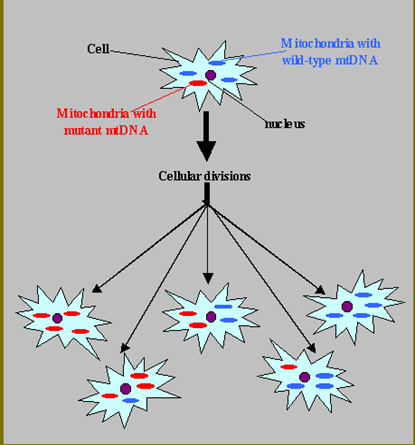
Threshold Effect.
energy requirements vary between tissues
mtDNA mutation burden varies tissue to tissue (heteroplasmy)
Tissue specific % mutant mtDNA threshold for disease
Phenotypic variability results
Example: Brain and Muscle have a lower threshold than Skin and Kidney
Mitochondrial Disorders: Presentation
Can present in almost any way and vary from person to person, but 3 general categories
“Classic” Mitochondrial diseases: reproducible, multi-organ pattern
Unexplained multi-organ dysfunction:
Hearing loss short stature
Diabetes + hypertrophic cardio myopathy
ophthalmoplegia +ptosis
Unexplained single organ syndrome: just hearing loss, epilepsy, GI
Often elevated Lactic acid in Blood or CNA and Mitochondrial proliferation in muscle
Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke like episodes (MELAS)
Age of Onset: before 40yo (average 5-15)
Clinical
Stroke like episodes + Epilepsy, Dementia
Muscle weakness (myopathy), Cardiomyopathy, Lactic Acidosis
Hearing-Loss, Retinopathy, Diabetes
CT/MRI: Infarcts→ but not seen in vasuclar regions: infarct occurs due to region engery insufficney from Mitocondrial
Etiology: heterogeneous mtDNA mutations (Often mt-t RNA) → VERY dependent on Heteroplasmy with individual
Myoclonic Epilepsy with Ragged Red Fibers (MERRF)
Adolescent onset
Clinical manifestations
Epilepsy (myoclonic)
Muscle weakness (myopathy), Lactic acidosis, Ataxia
Encephalopathy, Hearing Loss
EMG
EEG:
Muscle Biopsy: (if done on affected muscle) will show ‘ragged red fibers’ caused by mitochondria proliferation
Etiology: Single mtDNA-tRNA mutation 80 to 90%
Leber’s Hereditary Optic Neuropathy (LHON)
Age of onset 20-24 yo
Clinical:
Acute or sub-acute bilateral central vision loss→ Rapid progression to blindness (usually confined to optic nerve)
Rarely: heart block, dystonia, MS-like symptoms
Fundoscopy: early tortuous retinal arteries, followed by optic atrophy
Etiology: 95% mtDNA “ND” (electron transport subunit) gene mutations MATERNAL INHERITANCE
****4:1 M:F ration → X-linked modifier genes that make females less affected****
Chronic Progressive Ophthalmoplegia (CPEO)
Chronic Progressive Ophthalmoplegia (CPEO)
External ophthalmoplegia (eye weakness) → can’t look in certain directions
bilateral ptosis (eyelid drooping)
mild myopathy (limb weakness)
Onset ***AFTER*** 20yo (slowly progressive)
Etiology:
Mainly mtDNA deletions→ can be smaller or larger chunks of mtDNA (smaller =CPEO, larger=KSS)
Majority are SPONTEOUS
Subacute Necrotizing Encephalopathy (Leigh Syndrome)
Large spectrum of 75 genes (mito and nuc. but mostly nuc.) that cause an energy failure in the brain
6-12 months onset - death by 3-5 years (25% have later onset or slower forms)
Clinical: (often abrupt decompensations/regression with infection/fever)
Developmental ***REGRESSION***
Seizures, Ataxia, Hypotonia, spasticity
Ophthalmoplegia, Nystagmus, Optic atrophy
Diagnostic Testing
MRI: ***SYMETRIC LESIONS OF BASAL GANGLIA***
Elevated Lactic Acid in blood or Cerebral spinal fluid
10% mtDNA mutation
90% nDNA mutation
Lower % of mitochondria with the mutant mtDNA→ have NARP instead of Leigh (HETEROPLASMY)
Leigh Etiology
Genetic Heterogeneity
10-30% mitochondrial DNA mutations → maternal inheritance
90-70% nuclear DNA mutations → Classic Mendelian
HETEROPLASMY AFFECT: If a lower # of mito. in a cell have these mutations = Later onset Neuropathy, Ataxia, Retinitis Pigmentosa (NARP)
Pyruvate Dehydrogenase Complex (PDHC) Deficiency: Clinical + Testing
Failure to convert Pyruvate to Actyl-CoA (via PDH)
Lactic Acid levels elevated (***PDHC most common cause of Lactic Acidosis***)
Point mutation in NUCLEAR DNA
Clinical Features: Progressive intermittent neurologic deterioration
hypotonia, seizures, ataxia, ophthalmoplegia, dystonia
Presents similar to mitochondrial dysfunction
Suggestive Abnormal Tests
Plasma: increased Lactic Acid + Pyruvate, but normal ratio of Lactic Acid: Pyruvate
Distinguished from other Mitochondrial Disease: Pyruvate levels are NOT elevated
Cerebral Spinal Fluid: increased Lactic Acid
Pyruvate Dehydrogenase Complex (PDHC) Deficiency: Metabolism +Etiology
Failure to convert Pyruvate to Actyl-CoA (via PDH)
Lactic Acid levels elevated (PDHC most common cause of Lactic Acidosis)
Etiology: PDHC is a multisubunit complex
Catalytic components: E1, E2, E3
Regulatory component: PDH Phosphatase
Confirmation:
PDHC enzyme activity assay
Sequencing of
E1 → PDHA1 : MOST COMMON , X-Linked (males only)
E2 → DLAT, Recessive
Mitodoncrial Diease: Work Up
Serum levels: increased anion gap + metabolic acidosis
Lactic Acid: Pyruvate ratios (>30 Mito. Dis ; <10 PDHC Def.)
Imaging: brain MRI, Spectroscopy ( LA peaks over brain regions( BasalGang)
Basal Ganglia hypodensities: generalized atrphy
Hypoplastic corpus callosum if fetal lactic acidosis
Muscle Biopsy
Genetic Testing
Mitochondrial Disease: Muscle Biopsy
Allows for:
Detecting ragged red fibers (mito. proliferation)
Abnormal mitochondria proliferation
Detecting enzyme activity of the chain-genes
Mutational analysis of mitoDNA
Pitfalls:
need 1 gram of flesh (large amount)
biopsy of moderately affected muscle
may not distinguish exact genetic mechanisms
Genetic Testing
mtDNA:
Leigh Syndrome
LHON
MERRF (blood/muscle)
MELAS (blood/muscle)
NARP (blood/muscle)
KSS/CPEO (muscle)
nDNA (all in blood)
Leigh syndrome
MNGIE
Mohr-Tranebjaerg
Friedreich’s Ataxia
AR spastic paraparesis
AD PEO
Mitochondrial Disorders: Treatments
Less evidence for specific treatments that actually improve outcomes
Trials with Vitamins that optimize Electron Transport chain function:
Carnitine
Biotin
thiamine
Riboflavin
High Fat/ Low Carb diet: low carb→ less glycolysis→ less LA
Avoid Mito toxic meds
Reduce LA, control acidosis (dialysis/vent)
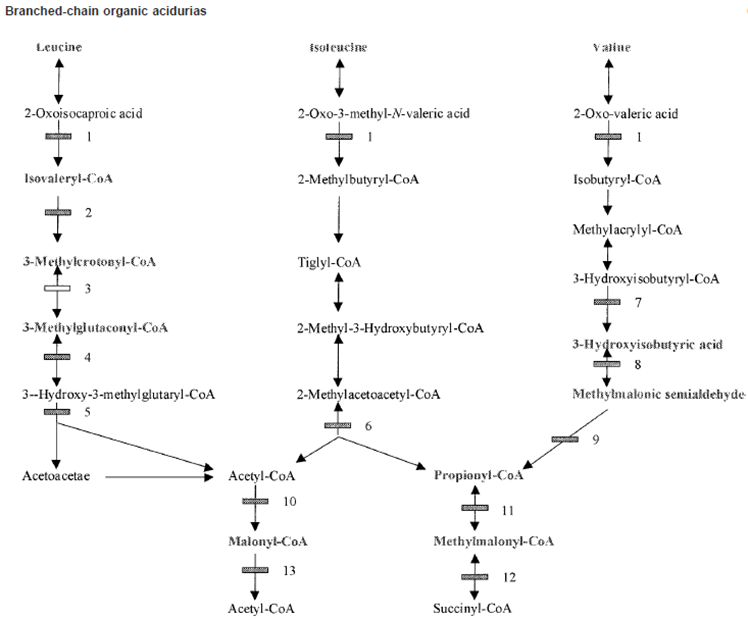
Organic Acidemias Background
Primarily disorders of Amino Acid Catabolism: Mainly
Branch Chain Amino Acids (BCAA)
Lysine
Toxicity comes from accumulation of ORGNIC ACIDS not from an A.A. acid accumulating
Causes metabolic acidosis with increased “Anion Gap”: Decrease in main anion Bicarbonate (HCO3-)
Secondary toxic effects of acidosis
Mitochondria→ Lactic acidemia
Urea Cycle → Hyperammonemia
Bone marrow→ Bone marrow suppression
CNS function→ Encephalopathy/Mental retardation
Major Presentations: Neonatal encephalopathic acidosis, late chronic/intermediate
All autosomal recessive
Metabolic Acidosis
Blood pH low due to excess acid (H+) vs Base (HCO3-)
normal range pH 7.3-7.45 (measure via Atrial Blood Gas)
Normal HCo3- level: 22-26 mEq/L
Mutiple etiologies for Metabolic Acidosis
Lowered HCo3- : loss through GI (diahrria), Renal tubule acidosis, Medications
Elevated H+: creation of abnormal acids in blood due to starvation, diabetes; Lactic acidosis due to mitochondrial dysfunction, Organic Acidosis
Clinical Consequences
Neonatal: non-specfic, similar to UCDs presenations,
Lethargy, vomting, Tachypena, Hypotonia, Seizures, Coma, Death
Adult: Devleopmental Delay, Ataxia, Neurological Deficits, (then the neonatal presenations)
Organic Acidemias
Newborn Screening detects many Organic Acidemias
Organic Acidemias Treatment
Restrict Dietary Protein disease specific amino acid free formulas
Prevent Catabolism provide sufficient protein free calories
Reverse Acidosis ± Hyperammonemia
Hemodialysis
Ammonia and lactic acid scavengers
Sodium bicarbonate, sodium benzoate, phenylbutyrate
Cofactor therapy for specific Disorders
Propionic Acidemia Metabolism
Failure of Propionyl-CoA carboxylase
Step 11 of Isoleucine and Valine metabolism:
Propionyl-CoA → Methylmalonyl-CoA via Propionyl-CoA carboxylase activity
Propinoyl-CoA: the activated mitochondrial form
Propionic Acid: free organic acid (interferes with NH3 removal, other stuff)
Propinolycarnatine (C3 - what NBS measures): the ‘detox’ / transport form that excess P-CoA gets converted to so it DOESN'T become Propionic Acid
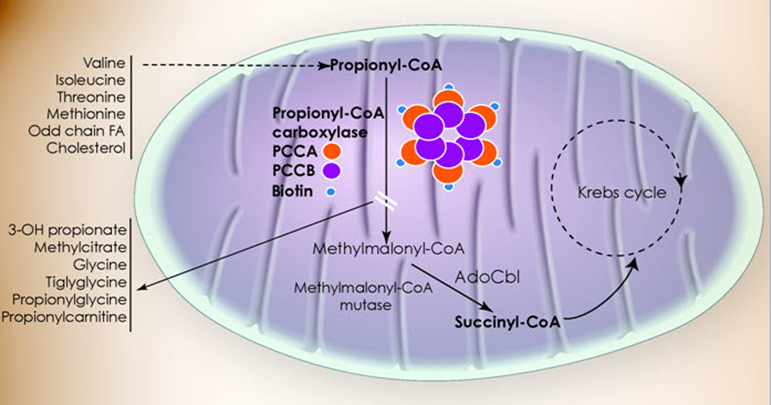
Propionic Acidemia
Also known as Ketonic hyperglycemia: high level of glycine and ketone bodies
Autosomal Recessive
Incidence 1:100,00 (higher in Saudia Arabia and Inuit)
Genetic Defect
Propinyl-CoA Carboxylase (PCC) alpha or beta subunit genes
some genotype/phenotype correlation (null alleles/deletions more severe)
Biotin cofactor for PCC
PA accumulation due to PA production from
MET/THR/VAL/ISO catabolism,
gut bacteria,
odd chain FAs
Untreated Propionic Acidemia
Classical Neonatal Encephalopathic Form
Normal at birth
Within a few days
Poor feeding, lethargy, vomiting hypotonia →encephalopathy, seizures, coma, death
Late-Onset Form
Developmental delays/regression
cyclic vomiting
protein intolerance
growth impairment
hypotonia
metabolic basal ganglia stroke
cardiomyopathy
Acute episode of toxic encephalopathy
Rare Cardiac Subtype isolated cardiomyopathy
Diagnosing Propionic Acidosis (PA)
Newborn Screening
Elevated Propionyl Acylcarnitine and ratio to other carnitine species (Propionyl-CoA gets combined with Carnatine to try buffer high Prop-CoA levels)
other etiologies: Methylmalonic Acidemia, Cobalamin Defects, Maternal B12 Deficiency, False +
Confirmatory Testing
Atrial Blood Gas: Elevated ammonia, low glucose, high acidosis, increased anion gap
Complete blood count: suppression of bone marrow→ less blood cells
Urine Organic Acid Analysis: High 3-OH-proprionate, mthylcitrate, tigly/proprionylglycine but NOT MMA
Plasma Amino Acid profile:
elevated glycine + glutamine, not homocysteine (seen with Cobalamin defects)
Acyl-Carnitine Profile: Elevated C3 acylcarnitine, not C4-DC unless SUCLA2 deficiency
PCC enzyme activity: can measure PC enzyme in leukocytes or fibroblasts
PCC Genotyping
Gene sequencing w del/dup analysis (99% detection rate)
Treating Propionic Acidosis
Acute Acidotic Encephalopathy
Remove acids and ammonia hemodialysis
severe hyperammonia: ammonia scavengers
Reduce PA production Protein restriction 24-25hr
Prevent catabolism: glucose and lipids IV
Enhance PA excretion: IV Carnitine
Decreased PA production in Gut: Antibiotics (Metronidzole
Biotin:
Chronic Treatment
protein restriction and MTVI-free metabolic formula
Oral Carantine, Biotin, and Antibiotics
Avoid decompensation
unresponsive to Tx → liver transplantation
Propionic Acidemia Deficiency Outcome
Treamtnet improves surivial, but invariable there is an affect to some degree
Neurodevleopmatl disabilty
metabolic basal ganglia stroke
seiures
pancreatisis
cardiomyopathy
gorwth impairment
nuetorpnia, AA defience
renal failure
premature ovarian fialure
hearing and vidual defecits (optic nerve atrphy)
Propionic Acidemia Deficiency Screening
Carrier Screening:
PRenatal Diaongis:
amontic fluid orgnaic acid measurment possible (some false negatives)
Methylmalonic Acidemia Pathway
Isoleucine and Valine
Methlymalonyl-CoA → Succinyl CoA via Methylmalonic-CoA mutase activity
Methlymalonyl-CoA accumulates
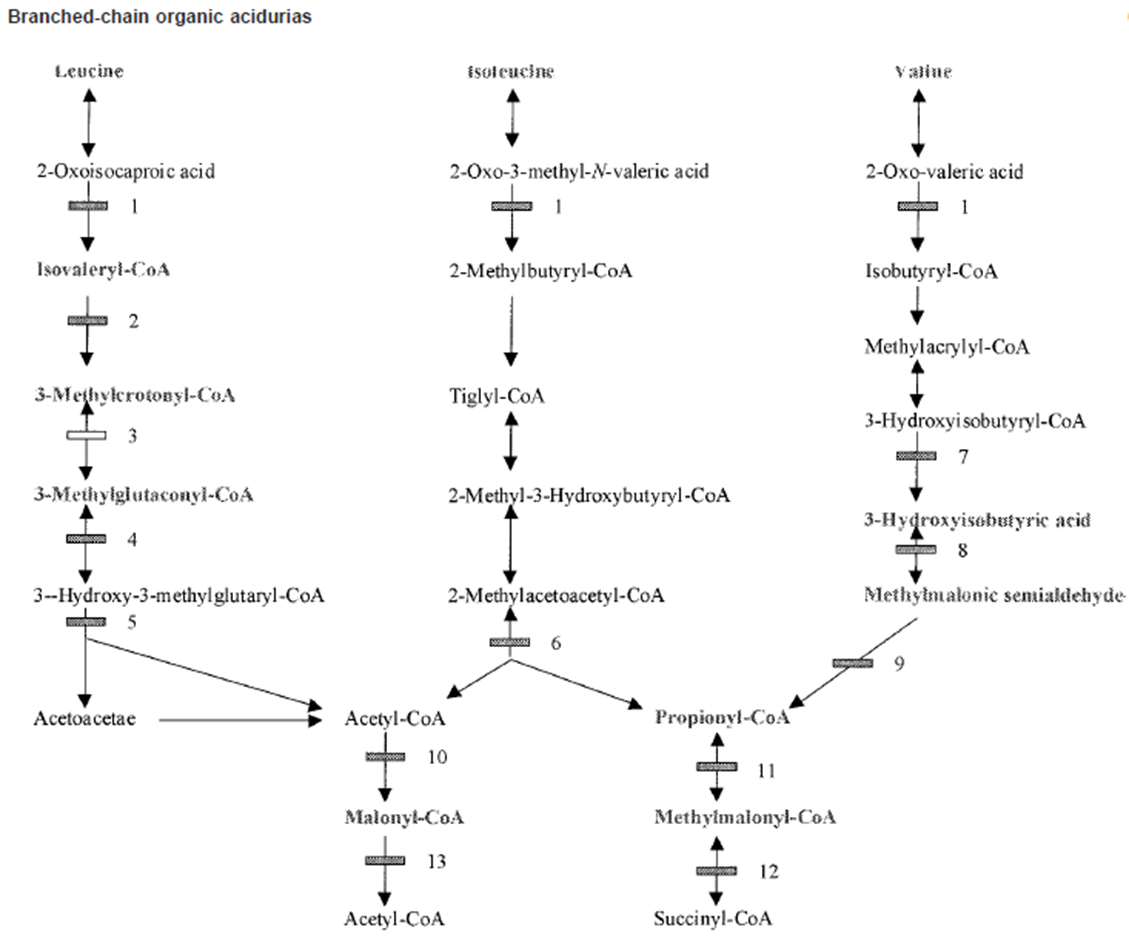
Methylmalonic Acidemia
Increased Methylmalonic Acid but not homocysteine (other forms of MMA have elevated homocysteine→ not primary MMA, but related to Adenosyl Cobalamin - A )
Genetic Defect: mutation of multiple genes cause similar phenotype
60% Methlymalonyl-Co mutase gene mutation (MUT)
37% Cobalamin A,B,D2 (MMAA, MMAB, MMADHC)→ the upstream vitamins that will be converted into Adenosyl Cobalamin→ leads to dysfunctional MM-Co mutase
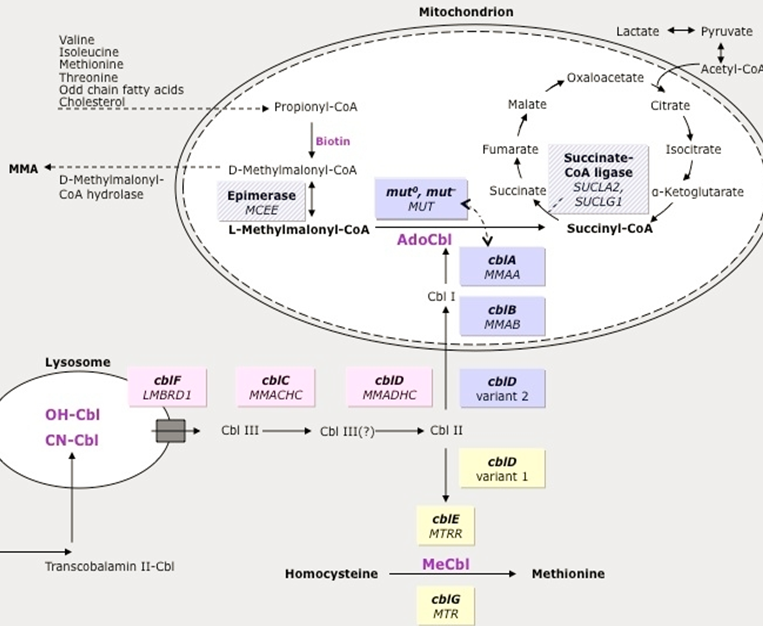
Untreated Methylmolaynic Acdiemia
Infantile Subtype: Most common mut0, cblB mutations
Normal at Birth
Within days to weeks: poor feeding, lethargy, vomiting, hypotonia, encephalopathy→ progress to seizures, coma, death
Intermediate phenotype: mut-, cblA, cblD2
Normal for month to years: fialure to thrive, devleopmental delay, hypotonia, poriten aversion→ risk of carastrophic decompensations
Benign Adult form: typically asymptomatic, can decomapnste
Diagnosing Methylmolaynic Acdiemia
Newborn screening: Elevated Propinoyl Acylcarnitine (and ratios) → but non specific
Confirmatory testing
Atrial Blood Gas, Ammonia Levels, Completel blood ocunt:
Hi AG metabolic acidsosi
Elevated ammonia
Low gluclose
pancytopenia
Urine Organic Acid: High MMA
Plasma Amino Acid profile: high glycine + glutamine, no Homocystine (Hcy)
CblC/D/F - Hcf + MMA high ;
cblD2/E/G - Just hcf High
Enzyme activity: fibroblasts
Genotyping on genes = 95%
Treating Methylmolonic Acidemia
Treat acute acidotic encephalopathy
Remove acids+amonia: hemodialysis
Reduce MMA production: protein restriction
Prevent catabolism: IV glucose and lipids
Severe hyperammonemia: Amonia scavengers
Decrease gut bacteria: Antibotics
HYDOXYCOBALAMIN (B12) injects: cofactor
Chronic Treatment
protien restriciton and MTVI-free meatolibc fomumal
L-Carnitine + OH-B12
Avoid decompensation
Methylmalonic Acidemia Treatment outcome
Most patient will have some degree of mental impairment, long term affects
Methylmalonic Acidemia Diagnosis
Prenatal/Preimplantation:
Ammonitic organic acid fluid analysis possible
Enzyme activity of CVS and amniocentesis
Iso-valeric Acidemia
issues with the Isovaleryl-CoA dehygroenase enzyme
LEUCINE PATHWAY ONLY
Build up of Isovalryl-CoA (Isovaleric Acid)
Disorder of Leucine metabolism only (unlike PA or MMA)
Genetic Defect
IsoValeryl-CoA Dehydrogenase (IVD) Gene Mutation
results in increased Isovaleric Acid
Sweaty feet odoer is prominent
Untreated Isovaleric Acidemia
Severe Neonatal Onset form
Normal at birth
Within day: poor feeding, lethargy, hypotonia, Sweaty feet order → encephalopathy, seizure, coma, death
Mid/Late Onset Form
unexplained failure to thrive and developmental delay
Benign Adult Form: Typically, asymptomatic but can mildly decompensate
Diagnosing Isovaleric Acidemia
Newborn screening: elevated Isovalerylcarnitine (C5 acylcarnitine) - used to ‘buffer’ Isovaleric Acid that builds up when Acylvaleryl-CoA builds up
Confirmatory testing
Blood tests:
High ammonia
Low glucose
High metabolic acidosis
Urine organic acid
High IVA
High isovaleryl glycine
Plasma AA levels:
High glycine
High glutamine
Enzyme activity: Fibroblast
Genotyping: exact genes unknown
Treating Isovaleric Acidemia
Treat Acute Acidotic encephalopathy
Remove acids and ammonia: hemodialysis
Reduce IVA production: protein restriction 24-26hrs
Prevent Catabolism: IV glucose and lipids
Enhance IVA excretion: IV carnitine
If hyper ammonia: ammonia scavengers
GLYCINE SUPPLMENTAITON-BINDS IVA
Chronic Treatment
Protein rection and LEUCINE-free metabolic formula
Oral L-Carnitine and L-Glycine
Avoid decompensation
Isovaleric Acdiemia Otucome
Outlook with Treatment is one of the best if treatment done early and effecetively enough
can be comepltely asymptomatic as long condition is monitored
Leucine tolerance gets better with age
Even if diaognsis is after neonatal period, and evne with major encaplapthic event in neonatal period—> longer term out look is vairable : CAN BE OK
Isovaleric Acidemia Prenatal Diagnosis
Ammniotic fluid can be checked for organic acids
Biotinidase Deficiency Pathway
Biotin is a vital cofactor for of number of different enzymes:
ALL ARE CARBOXYLASES
3-Methylcrontoyl-CoA carboxylase (Leucine)
Propinoyl-CoA Carboxylase (Isoleucine and Valine)
Malonyl-CoA decarboxylase
When there are mutations in the BIOTINADASE gene: Biotin is not properly recycled → these blocks develop
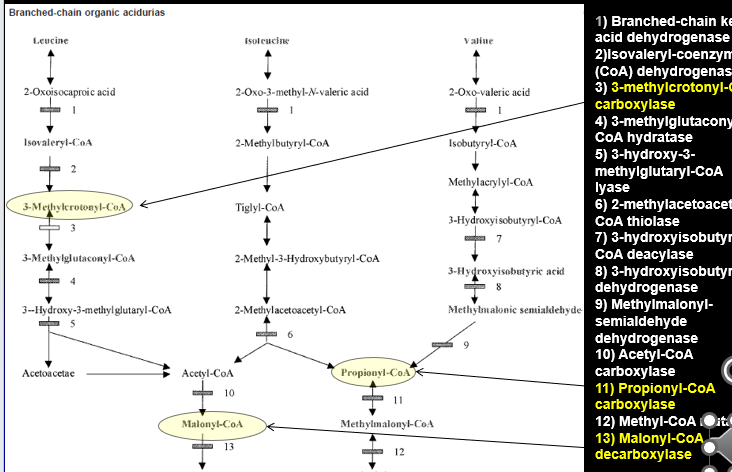
Biotinidase Deficiency (BTD)
Late multiple Carboxylase Deficiency
Slightly increased incidence in Hispanic and Middle Easter
Gene Defect: Biotinidase (BTD) gene
failure to recycle biotin = biotin deficiney
Biotin co-factor for the carboxylases: cannot combine and make function enzyme
Untreated Biotinidase Deficiency
Affect depends on the residual enzymatic activity when biotin absent
Profound Deficiency (<10% enzyme)
Normal at birth
Symptoms develop after few months
Developmental delay, seizures, hypotonia, ataxia, hearing loss, visual problems, ***alopecia***, ***eczema*** (unique to BTD)
Partial Deficiency (10-30% enzyme)
intermittent symptoms with stress
Symptoms can be irreversible once present
Diagnosing Biotinadase
Newborn Screening: Elevated C5-OH Acylcarnitine, but not specfic to BTD
Confirmatory Testing
Blood:
High ammonia
High acidosis
Low gluclose
Urine Organic Acids: multiple organic acids b/c Bitonaisde affects multiple enzymes —> refered to as ‘Multiple Carboxylase Defeicieny’ (MCD) on uOA
( )
Elevated Hydroxy-Isovalyrl-carnatine (C5-OH)
Enzyme activity: IMPORTANT STEP: blood sample
If Biotinadase activity is normal→ then issue is probably Holocarboxylase Deficiency (presents the same way but is earlier)
Genotyping: sequencing 99% detection
Treating Biotinadase Deficeincy
Rarely severyl acidotic or hyperammonemic
may occasionally need sodium bi-cabonate (adress acidty)
may occasionally need amonia scavnerge (adress amonia levels)
Insitute Biotin therapy immediately
Chronic treatment
Biotin
No protien restction
Avoid raw egg whites (has protein that binds Biotin)
Biotinadase Deficiency outcome
Extremely great outlook for patients (one of the best for Organic Acidemias)
As long as treatment is implemented BEFORE the development of severe symptoms
If detected after symptoms, some are irreversible: optic atrophy, hearing loss, developmental delay can presist
Biotinadase Deficiency Prenatal diagonsis
Biotinadase enzyme activity can also be measuredin the amniocytes and the amniotic fluid
Glutaric Acidemia Type 1 (GA1) Metabolism
NOT a Branch Chain Amino Acid metabolism disorder
Breakdown of LYSINE and TRYPTOPHANE
Lysine + Tryptophane → Alpha ketoadipic→ Glutyrl-Coa
Glutyrl-Coa→ Glutaconyl-CoA (shunt to Glutaontic Acid) via Glutaryl-CoA Dehydrogenase activity
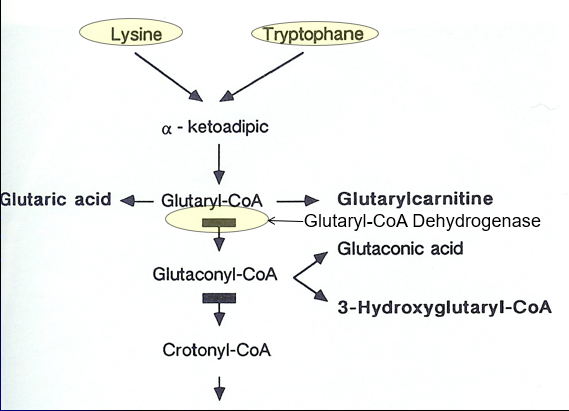
Glutaric Acidemia Type 1 (GA1)
“Cerebral” Organic Acidemia: Often normal
Genetic Defect: Glutaryl-CoA Dehydrogenase (GCDH) gene mutation causing defective Lysine + Tyrptohan metabolism
Glutaric Acidemia Type 1 (GA1) Symptoms
Often normal at birth or only macrocephalic
symptoms often begin prior to 2 years of age
May start with a Sudden neurologic decompensation: 75% by 14months—> fever, illness, metabolic stress
Primary symptoms
Stress-induced encephalopathy
Ataxia
Epilepsy
Myoclonus
Storke-like episodes
Glutaric Acidemia Type 1 (GA1) Diagnosis
Newboarn screening: Elevated C5-DC (glutaryl) Acylcarnitine
many False negatives
Confirmatory Testing
Blood: elvated ammonia, low gluclose, Aciditiy
Plasma + Urine: C5-DC glutaryl acylcarnitine + glutaric acid
Enzyme activity: fibroblast
CT/MRI: Cerberallar atrophy, basal ganlia infact and hemorrhage
Genotyping
Glutaric Acidemia Type 1 (GA1) Treatment
Reverse/Prevent Catabolism When sick: protien free calroeis during metaoblic stress
Dietary Mdofication
Low LYSINE and TRYOPTHAN
MEdicaitons
B2 (Riboflavin) is a COFATOR
Carntine: Binds Glutaric acid and remvoes it
Avoid Valproate (Bind Carnitine)
The Urea Cycle
6 Major Enzymatic reactions that occur in the liver within the Hepatocytes: the mitochondria + cytoplasm
2 Major functions
removal of nitrogenous waste (produced mainly from protein catabolism) → Ammonia incorporated into Urea for Excretion
Synthesis of amino acids: Arginine, Ornithine and Citrulline (become ESSENTIAL A.A. in deficiencies of the UREA CYCLE ENZYMES)
Major presentations of Urea Cycle Disorders:
Severe Neonatal Hyperammonemic encephalopathy (exception: Argine deficiency + late/mild onset variants)
All Autosomal Recessive except for Ornithine transcarboxylase deficiency (OTC):
OTC is X-Linked Recessive → only affects Males
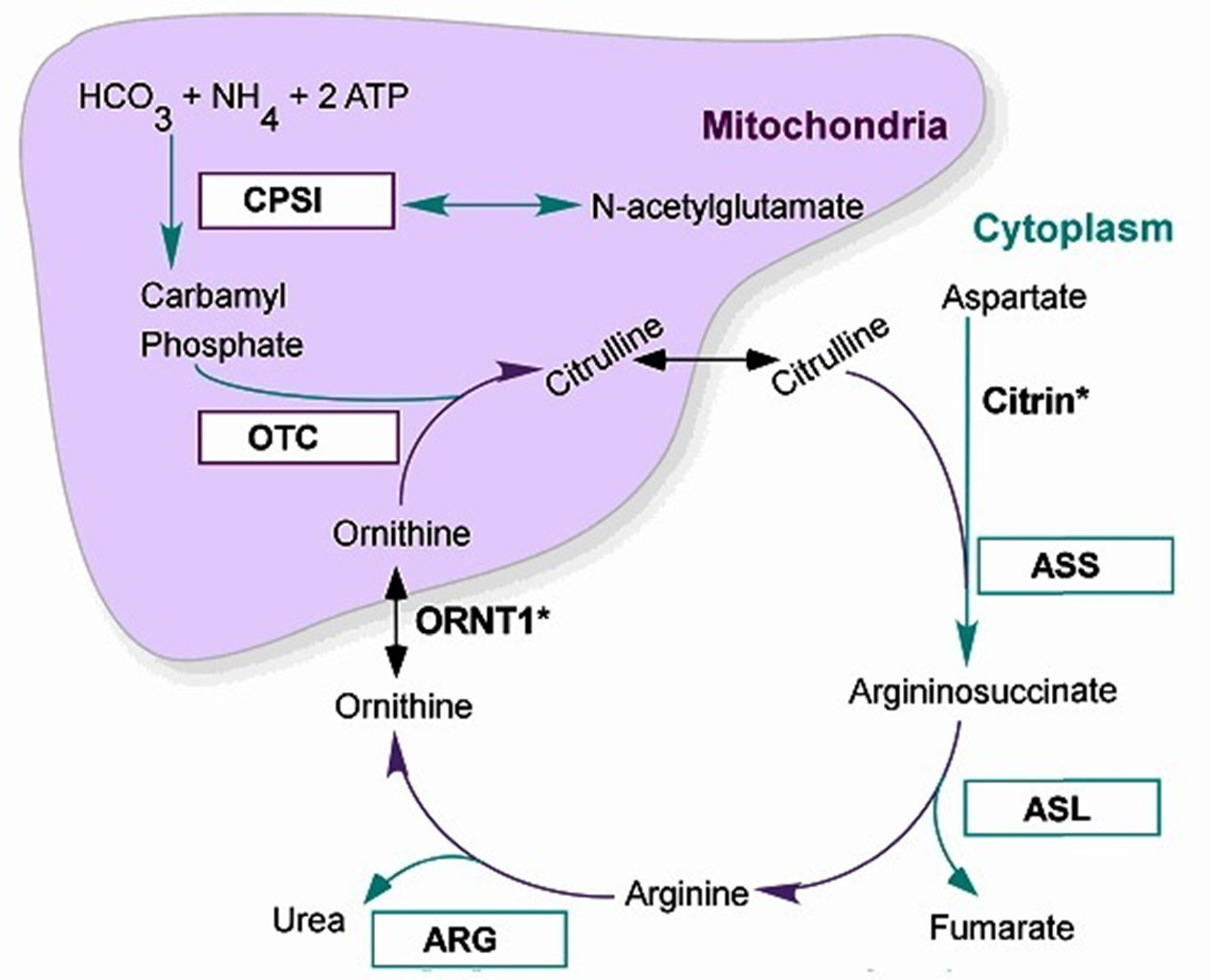
The 6 Urea Cycle Disrders
Correspond to the 6 steps of the Urea Metabolic cycle: taken individual relatively uncommon, but all together 1:8-35,000
N-Acetyl Glutamate Synthetase Deficiency
Carbamoyl Phosphate Synthetase (CPS1) Deficiency
****Ornithine Transcarbamylase (OTC) Deficiency****—> *****MOST COMMON*****
Arginosuccinic Acid Synthetase I (ASS1) Deficiency → also called Citrullinemia I
Arginosuccinic Acid Lyase (ASL) Deficiency
Arginase (ARG) Deficiency
Ammonia (NH3)
Ammonia is the product of the metabolism/catabolism of protiens/amino acids
Normal serum ammonia levels
Adults <35 mmol/L
Neonates <100 mmol/L (immature liver cells + increased tissue catabolism surrounding delivery)
Hyperammonemia: happen with great degree with IEMs of Urea Cycle But also seen with other metabolic disorders like Organic Acidemias (excess acid decreased Urea Cycle activity, lesser extent that UCDs)
Causes Neuronal excitotoxin increased extracellular glutamate +overexcitation of NMDA receptors→ Cell death and Cerebral Edema
Clinical Consequence
Acute severe elevation: seizures, coma, death
Mild chronic elevations: Brain atrophy, cognitive impairment
Classic UCD Presentation: Early
Neonatal Hyperammonemia Encephalopathy
In utero: protected my maternal urea Cyle activity of liver cells
At Birth in first 48hours: Ammonia levels rise quickly
Decreased feeding w/ vomiting
Lethargy
Tachypnea (rapid breathing)
Seizure activity
Followed by Rapid
Encephalopathy/Coma
Respiratory Failure
Cerebral Edema and Death
Late-Onset UCD Presentations
Variable age of onset and severity of Chronic and/or recurrence/Fluctuating symptoms:
Headache, Vomiting Ataxia and incoordination
Psychiatric/Behavioral disturbance: Delirium, ASD, ADD/ADHD, Manic episodes
Cognitive impairment: DD/MR, executive processing defects, early dementia
Often exacerbated/precipitated by:
Fever, Illness, fasting, post-partum, protein load (self restrict protein)
STILL AT RISK FORM HYPERAMMONEMC ENCEPHALOPATHY:
even if previously asymptomatic, can still be fatal
Mutiple Etiologies for Hyperammonemia
Things besides UCDs (MOST COMMON CAUSE) that can also cause the accumulation of excess ammonia:
Generalized Liver Deiasie (Acute or Chronic)
Non-genetic Causes: infections, Toxins, Trauma, Ischemia etc.
Genetic Causes
Non-IEM: Alpha-1 Antitrypsin (accumulation in liver=chrossis), Alagille syndrome etc
Non-UCD IEM
Aminoacidopathies: Tyrosinemia Type I
Organic Acidemia: elevated Lactic acid which inhibit NAGS
Primary Mitochondrial disorders
Fatty Acid oxidation Defects: Reye-Like syndrome
Carbohydrate Metabolic defects: Galactosemia, Fructosemia
Metal Processing defects WILSONS DIEASE, hemochromatosis
Primary UCDs
OTC Deficeiny Metabolism
X-Linked Recessive Form
Within the Mitochondria of Liver Cell:
Carbamyl Phosphate + Ornithine → Citrulline via Ornithine transcarbamylase (OTC) activity: a Block at OTC = decreased Citrulline, increased Arginine and Aringnosuccinate (along the cycle)
Ammonia + Orotic Acid also increased (run-off product of carbamyl phosphate NOT becoming citrulline, not usually accumlated)
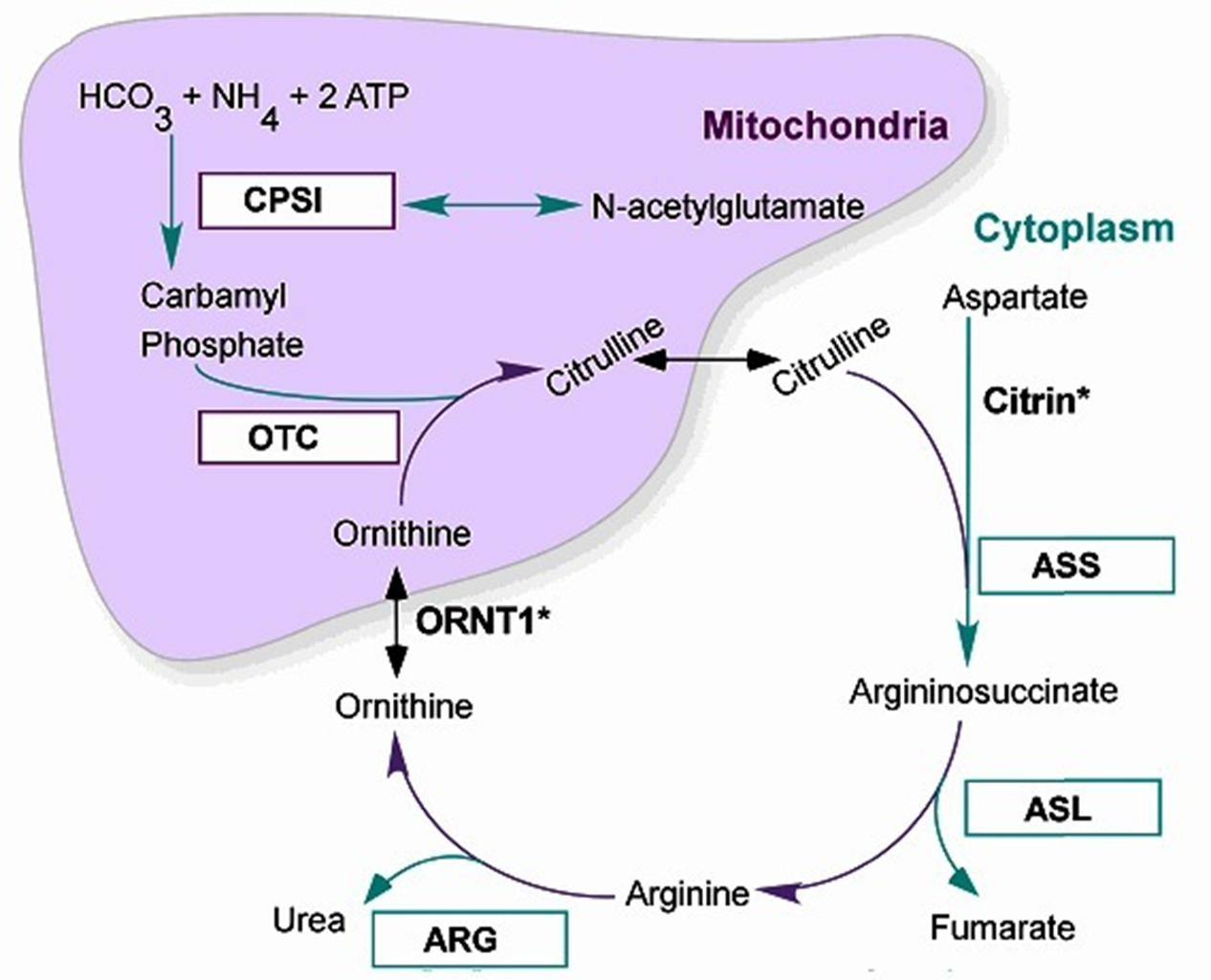
OTC Deficiency
Orthine Trancarbamylase (OTC) Deficiency:
Most Common UCD
X-Linked Recessive Inheritance
Incidence 1:56,000 (not increased in any specific group)
Genetic Defect: Orthine Transcarbamylase (OTC) Gene mutation
High Orotic Acid + Ammonia : Low Cirtrulline+ASS+ARGE
Female carriers most common, with symptomatic male probands
Non-carrier females: 3-4% germline mosaicism rate (apprecaible)
Untreated OTC Deficiency
Classic (usually males, only very rarely females)
At Birth to 24/48 Hours: Asymptomatic
2-3 Days old: Progressive hyperammonemia Encephalopathy
Poor feeding
Vomiting
Lethargy
Hyperventilation
Seizures
By 1 week Old: Lethal hyperammonemia Encephalopathy
Cerebral edema
Hypothermia
Coma
Respiratory failure
Death
MILD (Mild mutation males/symptomatic Females - 15%)
Episodic hyperammonemia Symptoms: ranging form mild to life-threatening
Chronic symptoms
Protein avoidance
recurrent headache
neuropsychiatric difficulty
Diagnosing OTC Deficiency
Newborn Screening: not done in all states (like NY) but ME, MAN,RI,VT
LOW CITRULLINE
Diagnostic Evaluation
Ammonia Level: symptomatic >100, Encephalitic >200
Blood gas/Metabolic profile/lactic acid levels: exclude other IEMs
Plasma Amino Acid Profile + Urine Organic Acid Profile
High glutamine/alanine + citrulline/ASA/ARG low in blood
High Orotic acid in urine
****Allopurinol given to female carriers can expose OTC by preserving Orotic Acid and show Orotic Acidemia****
Confirmatory Testing
Enzyme activity analysis: needs liver biopsy and not 100% in females due to x-inactivation in the liver
OTC genotyping via sequencing +del/dup analysis: 60-90% detection
Treating OTC Deficiency
Acute Encephalopathy treatment
Radpidly remove ammonia: hemodialysis/hemofiltration
Reduce production: protein cessation 12-24hours, decrease nitrogenous waste
Prevent Catabolism: high calorie, protein free - IV glucose + lipids and IVE arginine (become essential AA b/c it cannot be synthesized ‘like normal’)
Provide other ammonia removal agents: IV ammonia scavengers Sodium-Benzoate + Sodium-Phenylbutyrate
Chronic Treatment: after acute encephalopathy has resolved
Protein restriction + essential AA formula
Oral cituralline or arginine and ammonia scavenrs (Sodium-Benzoate + Sodium-Phenylbutyrate provide alternative pathways for ammonia excretion)
Avoid Valproic Acid, fasting, Fever, steroid, protein load
Liver transplant: only if neurologically intact after EPISODE + poor response to chronic treatment
OTC Deficiency Outcome
Outcome with Treatment: Variable
Neurocongtive delvepmn depens on intiala hyperammoneicm encephalyopth udration and control of subsquent amonia/glutamate leves
Ofente ID, ADHA, executive defcits, brain attrophy (even in midl males and symptomatic females)
OTC Carrier Screening + Prenatal Diag
Carrier Screening:
Molecuelar if mutation known in proband
Allopurinol Loading test show Orotic Acid elevations
Prenatal implantation: No enzyme/metabolite testing is useful, activity of OTC contained to liver cells
Fatty Acids
Under fasting conditions, fatty-acids are the stores used once 8-hour of carb derived glycogen is used up
Lipolysis: fatty acids get released into blood stream to be used by the body and converted into energy as Actyl-CoA and gluconeogenesis
Fatty acids can also produce ketone bodies via Bet-Oxidation to serve as energy for the BRAIN
Fatty acids:
3 acyl groups with attached glycerol group + carbon chain
Many fatty acids based upon the length of their carbon chain + the saturated double bonds
Most dietary fat stored as Palmaric acid (C16) or Stearic acid (C18)
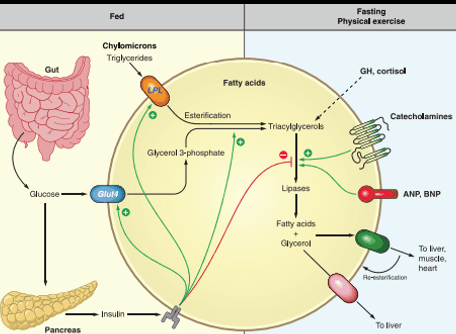
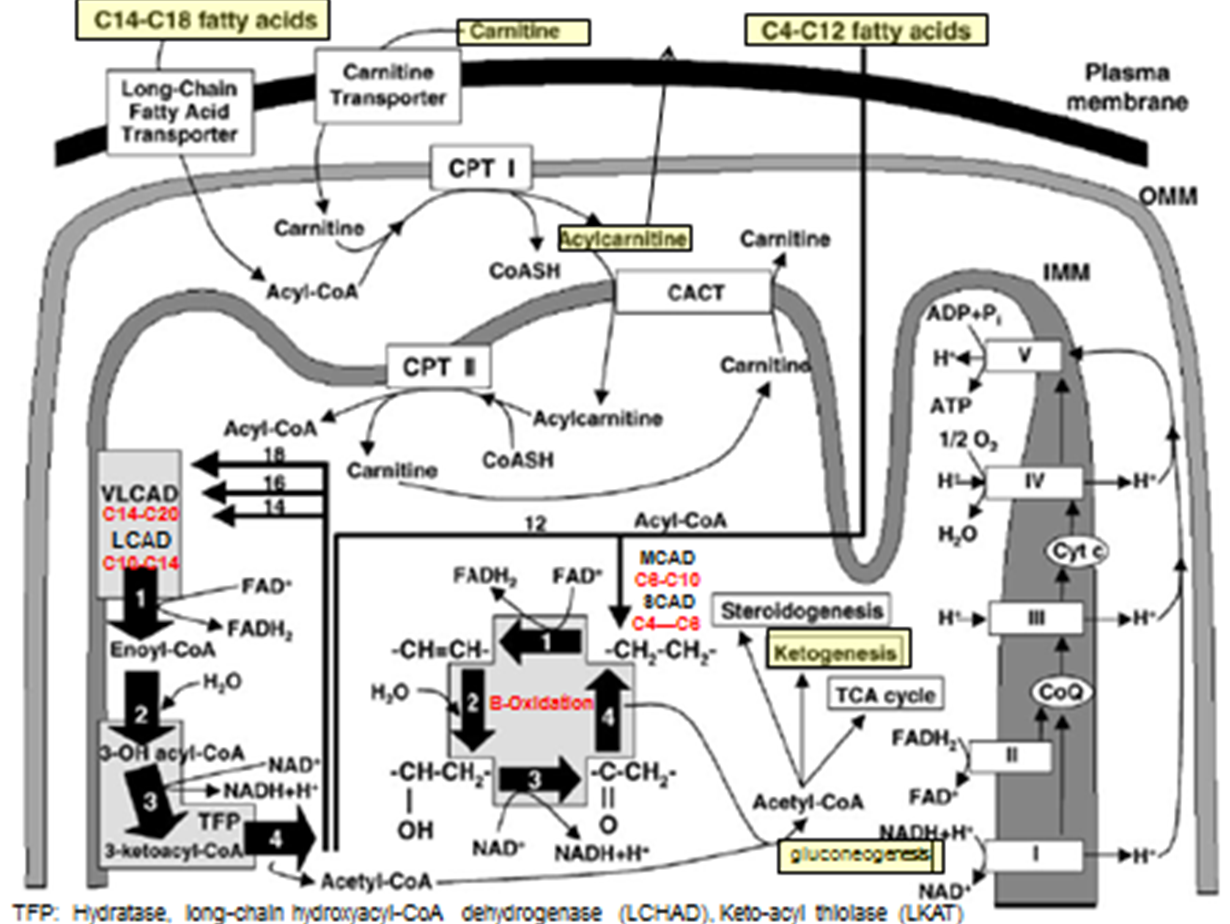
Fatty-Acid Metabolism Pathways: C14-C18 transport step
Fatty acids needs to be converted to Acetyl-CoA
C14-C18 Fatty acids → transport across plasma membrane via Long chain F.A transporter
Converted to Actyl-CoA (But cannot enter inner mitochondrial membrane)
Acyl-CoA + Carnitine → Acylcarnitine (of varying lengths) via CPT1 activity (TRANSPORT PREP STEP)
Acyl Carnatine moved into inner membrane in exchange free cranatine out via CACT activity (TRANSPORT STEP)
Acylycratine → Actyl CoA + Carnitine via CPTII activity (LIBERATION STEP)
Acyl-CoA can now enter into BETA OXIDATION
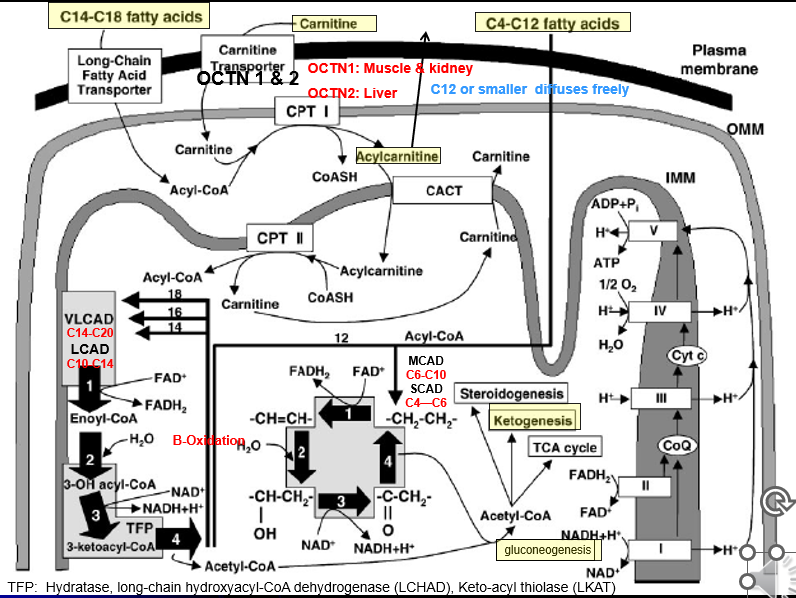
Fatty-Acid Metabolism Pathways: C14-C20 B-Ox
Special Cleavage Steps: Only done for fatty acidsC14 or longer
Acyl 2 carbon is cleaved from it via the Mitochondrial Trifunctional Protein
the Acyl-2C’s liberated can then undergo Beta-Oxiadtion
Each turn liberated a 2C Acytl CoA group
Fatty-Acid Metabolism Pathways: C4-C12 B-Ox
Smaller fatty acids can diffuse freely across all membrane of the cell + mitochondria: no need for the transporter or Beta-oxidation steps
Each turn liberated a 2C Acytl CoA group
Fatty Acid Oxidation Defects: Pathophysiology
Defective utilization of Fatty Acids (Acyl groups) for energy
Rapid glycogen depletion→ hypoglycemia in fasting sate (can’t switch over to FA B-Ox to maintain blood glucose levels)
Deficiency of energy substrate for muscles and brain
Muscles: F.A → Acetyl-CoA for energy
Brain: F.A → Acetyl-CoA → Ketone bodies (ketogenesis step)
Accumulation of unmetabolized F.A in liver and muscle
Liver disease
Myopathy
Cardiomyopathy
Fatty Acid Oxidation Defects: Major Clinical phenotypes
Hypoketotic + hypoglycemic
Glucose levels drop quickly, Ketone bodies made at low levels
Cognitive and developmental insults to the brain
Body TRIES to breakdown F.A., but can’t convert smaller chunks to the 2-Acetyl-CoA via B Ox
F.A.s liberated into blood stream = buildup within muscle and liver → dysfunction
Myopathy + Cardiomyopathy
Hepatic failure/Liver dysfunction (Reye Syndrome)
Maternal liver disease → affected fetus can create issue in the mother w F.A build up
Fatty Acid Oxidation Defects: Etiologies
Disease of the Carnitine pathway:
primary: defects in the protein pathways
secondary: nutrition deficiencies
Disease of Fatty Acid B-Oxidization
L-Carnitine Amino Acid
Binds to Acyl (organic +inorganic) residues (F.A) in the blood and enables transport into cells + their elimination → (why we supplement with Carnitine in Organic Acidemias: for their elimination w carnitine)
Long chain (>C12) needs it to get their long “acyl” chains into the inner mitochondria membranes where “Digestion” step + B-Ox can occur
Sources: we can make a-little, but not enough on its own
Diet: Milk + meat
Reabsorption: Kidneys via Caratine trnasporter protien
Carnitine Testing
Plasma Carnatine levles: Free, unbound and Acyl-Cartines (boudn to Fas and organic acids)
Plasma Acyl Carnatine prolei: quantity the diffrent types of bound-caratine resdiues
Fatty Acid Oxidation Defects: Key Concepts
All Autosomal Recessive
Spectrum of Severity
Phenotypes vary but always have
Hypoketotic + Hypoglycemia
Myopathy and/or Cardiomyopathy
Liver failure (Reye Syndrome)
SCIDs can ocure fruently: overnight fast casues sudden hypoglycemi even that kills them
Fatty Acid Oxidation Defects: Therapy
Acute Illness/Fastine
IV Dextrose (immediate)
Carnitine (some FOADs): overdrive membrane import of FA and remove excess FAs
Monitor for
hypoglycemia
liver failure
Muscle breakdown
Chronic
Avoid prolonged fasting
frequent feedings: CORNSTARCH (McArdles?)
Supplment with Medium-Chain-Triglycrides (“MCT”) oil <C10 for some (not MCAD/SCAD) → doesn’t need transporter or Digestion steps before B-Ox
Supplment with L-Cartine for some (not LCHAD)
Avoid liver toxic or carantine lowerin medication (valproic acid, salicylates, some anethetics)
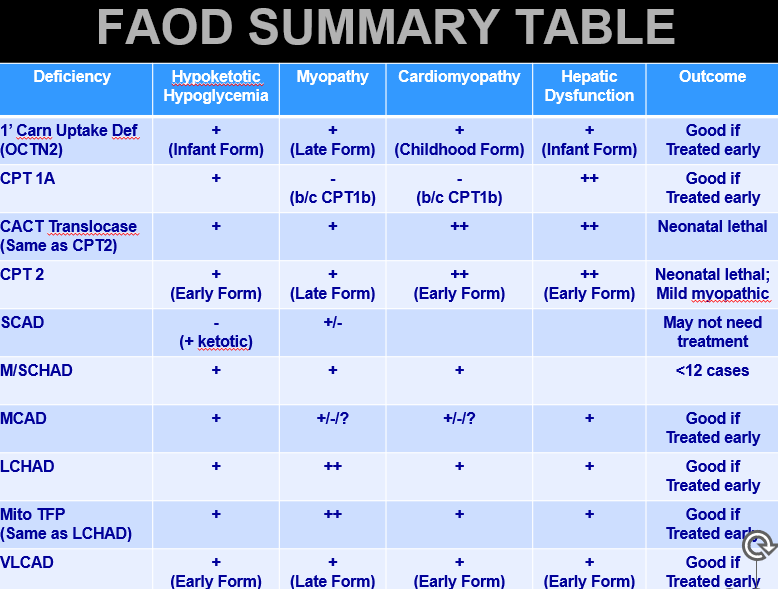
Overview of FAOD Clinical Issues
OCTN2 Deficiencey: Metabolism
OCTN1 and OCTN2 transport Carnitine across plasma membrane
OCTN1: Primarily in liver
OCTN2: Primarily in Muscle + Kidney
No OCTN2 = No carnitine = Long F.A.s don’t undergo B-Oxidation in the Muscle
Also: Kidney→ not enough Carnitine is re-absorbed and therefore too much excreted = very low levels of carnitine
OCTN2 Deficiency
Also called Primary Carnitine Uptake Defiencey
Etiology
SLC22A5 gene mutation→ reduced Carn. uptake by Kidney +muscle→ low blood and muscle Carn+ Acyl-Carn
Can also just be caused by LOW-Diet CARTNATINE
OCTN2 Deficiency: presentation
Forms: Severe Infantile, Mild Child/late Onset
Infancy:
Hypoketotic hypoglycemia
Liver failure
Child:
Myopathy + Cardio myopathy
Liver affects are less: when Carn is severe enough, B-Ox affected through whole body → but Child type, Carn is enough to avoid defieciny inthe liver but not the rest of the body
Late
mild myopathy or asymptomatic
OCTN2 Deficiency: Diagnosis
NBS: Low C0 and Acyl-Carnitine levels (Low level of ALL the species) → NEED TO TEST MATERNAL LEVELS AS WELL
Confirmation
Carnitine and Acyl-Carnitine levels
Enzyme activity: Fibroblasts
Molecular: finds 70%
OCTN2 Deficiency: Treatment and outcome
Treatment
high-dose Carn
Avoid fasting
IV Dextrose
Outcome
Good if treated before severe decompensation
Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficeny
VLCAD Drives the initial steps of fatty acid beta oxidation for the long chain fatty acids (>C14)
VLCAD Deficiency: Pathophysiology
ACADVL mutation → Deficient C14-20 B-oxidation
Generally more mild, later onset, exercise
VLCAD Def: Presentation
Infant:
Hypoketoitic hypoglycemia
Liver failure
myopathy
cardiomyopathy
Child: Cardio myopathy
Late: *******EXCERCISE MYOPATHY: WAY MORE COMMON FORM *****
VLCAD Def: Diagnosis
NBS: Elevated C14:1 ratio and longer chain Acyl-carnitine
Confirmation:
Carnatine and AC
Enzyme activity: Skin, white blood cell, aminotic fluid
Moelcualr: 85-93%
VLCAD Def: Treatment + Outcome
Treatment:
Avoid fasting with frequent feeding and IV dextrose when ill
Cornstarch
MCT Oil
+/- Carnitine
High-carb/low-fat diet
Outcome
good if treated before severe decompensation
Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency
Most common FAOD and the first to be added to NBS
the meatbolism of C8-C10 length acyl-CoA Fatty Acid groups
These can move freely acros the inner mitocondrial memebrane WITH OUT then eed for Carnatine complex (so no Acylcarantine needed
Once inside,C8-C10 Acyl-CoAs → *****BETA OXIDATION via MCAD****→ C4-C6 AcylCoAs
MCAD Deficiency = C8-C10 Buildup in blood and tissue + C8 Octanoyl Acycl Carainatase (Toxic)
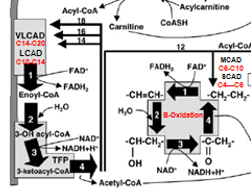
MCAD Def: Pathophysiolo
ACADM gene mutation → lowered C6-C10 Fatty acid B-Oxidation = build up of C8-C10 in tissues (C8 more prevlant vs C6 or C10)
Also alternative FA metablism pathway (Omega Oxidaiton) creates new species (dicarboxylic acid)
MCAD Def: Presentation:
Typical presentation at 3-24months old with Fasting/Illness
Hypoketotic hypoglycemic
Liver disease
Often Sudden Infant Death syndrome→ usually the first personation you will see (sleeping longer through the night, greater chance for fasting affects to kick in)
MCAD Def: Diagnosis
NBS: Elevated C8, lesser C6 and C10 : (C8 >C6/C10)
C8 (Octanoylcarnatine)
C6 (Hexanoylglycine)
Dicarboxylic Acid (Alternative FA Metabolsim Product)
Confirmation
Carnitine
AC
uAG
Enzyme activity: Skin, WBC, amino/CVS
Molecualr: 90%
MCAD Def: Treatment + Outcome
Treatment
Fasting with Frequent feeding and IV dextrose when ill
Cornstarch
+/- Carnitine
High carb/low-fat diet
****NO MCT OIL!!!!****
Outcome
Good if treated before severe decompensation
Maternal liver disease reported (AFLP/HELLP)