Medicinal plants post-mids
1/74
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
75 Terms
HPLC core idea
A liquid (the mobile phase) pushes your sample through a tube packed with solid particles (the stationary phase).
Different molecules interact differently with this solid packing, so they move at different speeds and come out at different times.
This difference in travel time allows separation.
Main components of an HLPC (solvent reservoid, pump, injector)
1. Solvent Reservoir
Holds the mobile phase (e.g., water, acetonitrile, methanol).
The solvent composition can be constant (isocratic) or gradually changed (gradient).
2. Pump
Pushes the mobile phase at very high pressure (typically 1,000–6,000 psi).
High pressure is needed because the column packing is very fine.
3. Injector
Introduces a precise, tiny volume of sample (usually 5–20 µL) into the flowing mobile phase.
This ensures reproducibility.
HPLC components (Column, detector, chromatogram)
4. Column
The “heart” of the system.
It contains tightly packed silica-based particles, often treated with different chemical groups (C18 is the most common).
Molecules interact with these particles depending on their polarity, hydrophobicity, charge, etc.
5. Detector
Measures molecules as they leave the column.
Common detectors:
UV-Vis (most common)
Fluorescence
Refractive index
Mass spectrometer (LC–MS setup)
The detector converts chemical presence into an electrical signal.
6. Chromatogram
A graph of detector response vs time.
Each peak represents a separated compound.
Retention time (t_R) helps identify compounds.
Peak area helps quantify them.
How separation happens in HPLC
Normal-phase HPLC
Stationary phase: polar
Mobile phase: nonpolar
Polar molecules stick longer → longer retention times.
Reverse-phase HPLC (most widely used)
Stationary phase: nonpolar (e.g., C18)
Mobile phase: polar (e.g., water + acetonitrile)
Nonpolar compounds stick longer.
Ion-exchange HPLC
Separates based on charge.
Size-exclusion HPLC
Separates based on molecular size; larger molecules elute first.
Why is high pressure needed in HLPC
2. Why High Pressure Is Needed
The column is packed with extremely small particles (1.7–5 µm).
Small particles give better separation but create high resistance to flow.
Pumps apply high pressure (up to several thousand psi) to push the mobile phase through the column.
Pressure improves resolution and reduces analysis time.
Explain differential retention (HPLC)
3. Core Principle: Differential Retention
Each compound interacts differently with:
The stationary phase (solid packing)
The mobile phase (solvent mixture)
Stronger interaction → slower movement → longer retention time
Weaker interaction → faster movement → shorter retention time
This selective retention is the basis of chromatographic separation.
Retention time (HPLC)
Retention time is the time between sample injection and the detection of that compound at the end of the column.
It is used to:
Identify compounds (qualitative use)
Compare samples across runs
Monitor consistency in method validation
Chromatogram (HPLC)
A chromatogram is a plot of detector signal versus time.
It displays:
Peaks representing each compound
Retention time values
Peak area (quantitative information)
Peak shape and resolution
The area under the peak is proportional to the amount of that analyte.
Resolution (HPLC)
Resolution describes how well two peaks are separated.
High resolution means:
Distinct peaks
Accurate quantification
No peak overlap
Resolution depends on:
Column efficiency
Selectivity
Mobile-phase strength
Particle size
Column length
Efficiency and plate theory (HPLC)
Efficiency describes how sharply a peak appears.
It is expressed as the number of theoretical plates (N).
Higher efficiency → narrow peaks → better separation.
The Van Deemter equation explains factors affecting efficiency:
Eddy diffusion
Longitudinal diffusion
Mass-transfer resistance
Smaller particle size and optimal flow rate improve efficiency.
Selectivity (HPLC)
Selectivity is the relative difference in retention between two analytes.
It is the most important factor for achieving separation.
It depends heavily on:
Stationary-phase chemistry
Mobile-phase composition
pH (for ionizable compounds)
Changing selectivity is often the first step in method development.
Isocratic vs Gradient Elution (HPLC)
Isocratic
Mobile-phase composition stays constant
Good for simple mixtures
Stable baseline
Shorter equilibration time
Gradient
Mobile-phase composition changes during the run (e.g., increasing organic percentage)
Required for complex mixtures
Improves separation of compounds with very different polarities
Achieves narrow peaks even for late-eluting analytes
Quantitative vs Qualitative Use
Qualitative
Identify compounds by retention time
Assess purity
Check for degradation products
Quantitative
Use peak area to determine concentration
Calibration curves needed
Strong reproducibility required
HPLC (Solvent reservoris)
2.1 Solvent Reservoirs Purpose
Hold the mobile phase solvents.
Key Points
Usually glass or solvent-resistant plastic bottles.
Labeled A, B, C, D for multi-solvent mixing systems.
Mobile phase must be filtered and degassed to prevent air bubbles.
Degassing methods include vacuum suction, helium sparging, membrane degassers.
Common Solvents
Water (HPLC grade)
Methanol
Acetonitrile
Isopropanol
Buffer solutions (phosphate, acetate, formate)
HPLC (pump)
Purpose
Pushes mobile phase through the system at high pressure.
Key Points
Maintains accurate, steady flow rate (0.1–2.0 mL/min typically).
Capable of producing 1000–6000 psi depending on column size and particle size.
Must deliver pulse-free flow for stable baselines.
Types of Pumps
Reciprocating pumps (most common)
Syringe pumps (low pulsation, precise)
Pneumatic pumps (older models)
HPLC (injector)
Purpose
Introduces a precise volume of sample into the mobile-phase flow.
Key Points
Injection volume typically 5–20 µL.
Must be highly reproducible for quantitative work.
Types
Manual injector (rotary Rheodyne valve): operator turns a handle to inject.
Autosampler: robotic arm injects many samples automatically.
Sample Loop
A fixed loop (e.g., 20 µL) fills with sample before injection.
Ensures consistent sample volume.
HPLC (Column)
Purpose
The site where separation occurs.
Key Points
Stainless steel tube packed with silica-based or polymeric particles.
Typical dimensions:
Length: 50–250 mm
Internal diameter: 2–4.6 mm
Particle size: 1.7–5 µm
Stationary Phase Types
Reverse-phase (RP): C18, C8, phenyl
Normal-phase: bare silica, amino, cyano
Ion-exchange: sulfonic (cation), quaternary amine (anion)
Size-exclusion: porous beads sorted by size
Chiral phases: for enantiomeric separation
Guard Column
A small sacrificial column placed before the main column to protect it from particulates and degraded compounds.
HPLC (detectors)
Purpose
Detect and measure analytes as they exit the column.
Common Detectors
UV–Vis detector
Most commonly used
Suitable for compounds with chromophores
Photodiode array (PDA/DAD)
Measures full UV spectrum
Detects peak purity and identity
Fluorescence detector
High sensitivity for fluorescent compounds
Refractive index (RI) detector
Universal detector
Not useful for gradient elution
Evaporative light scattering detector (ELSD)
For non-volatile compounds lacking UV absorption
MS detector (LC-MS)
Extremely sensitive
Provides molecular weight and fragmentation
HPLC (chromatography software)
Purpose
Records the detector signal and converts it into a chromatogram.
Functions
Peak integration
Peak area calculation
Calibration curve generation
Retention time tracking
Method control (pump settings, gradient programming, detector wavelength)
HPLC (connecting tubings and fittings)
Purpose
Carry mobile phase and sample between modules without leaks.
Key Features
Stainless steel or PEEK tubing
Internal diameters selected to reduce dispersion
Finger-tight fittings or high-pressure ferrules
HPLC (Waste management system)
Purpose
Collects waste solvent safely.
Key Points
Must be compatible with organic solvents
Includes vapor traps to reduce exposure
Prevents pressure buildup in waste bottles

HPLC diagram
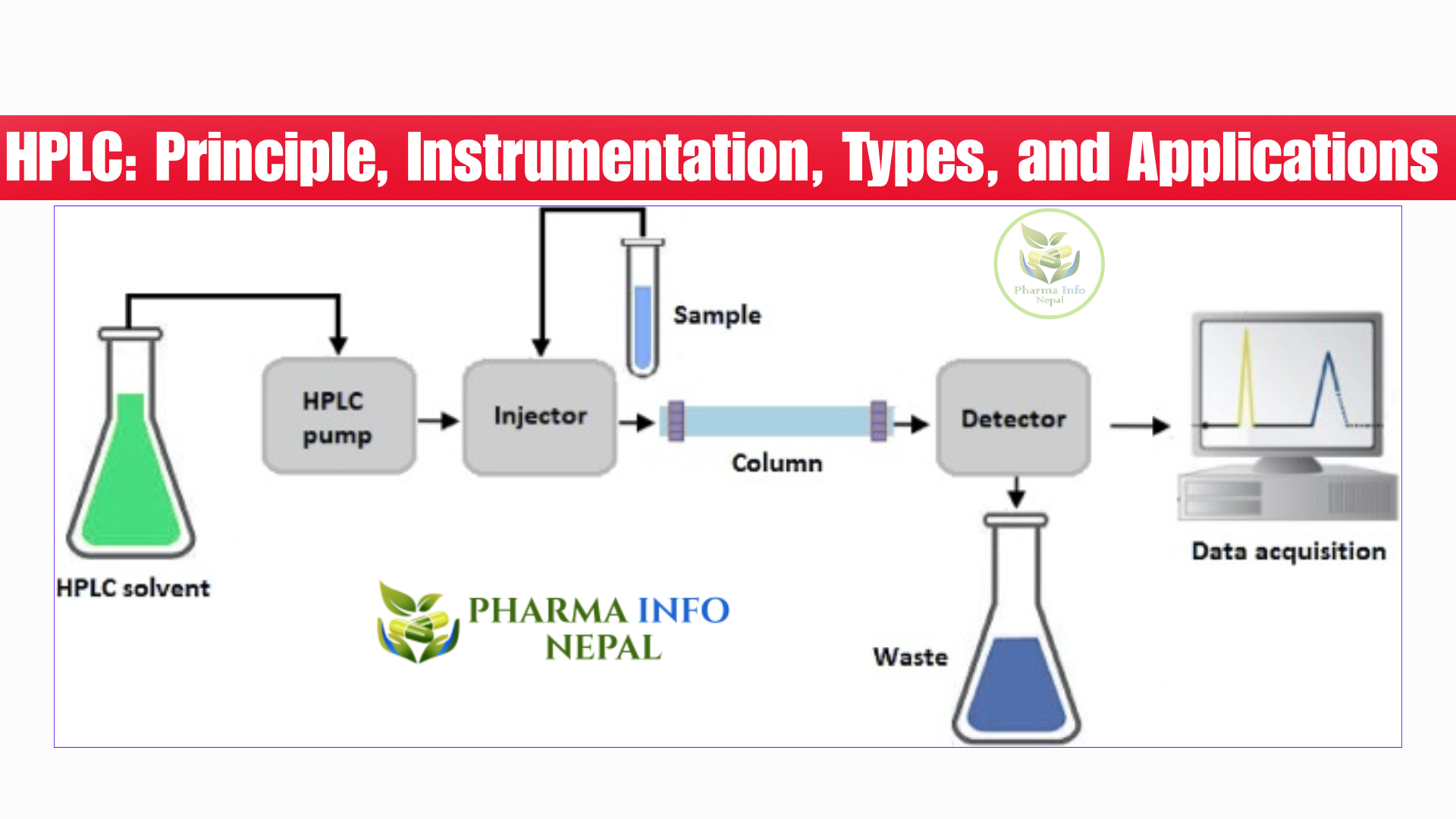
Reverse phase HPLC
3.1 Reverse-Phase HPLC (RP-HPLC)
Most widely used form of HPLC.
Principle
Stationary phase: nonpolar (hydrophobic), usually C18, C8, phenyl.
Mobile phase: polar (water + methanol/acetonitrile).
Separation occurs by hydrophobic interactions.
Behavior
Nonpolar compounds interact strongly → elute later.
Polar compounds interact weakly → elute earlier.
Applications
Pharmaceuticals
Natural products
Metabolites
Peptides
Most small organic molecules
Normal phase HPLC
Opposite polarity pattern compared to reverse-phase.
Principle
Stationary phase: polar (silica, amino, cyano).
Mobile phase: nonpolar (hexane, chloroform, isooctane).
Separation based on polar interactions.
Behavior
Polar compounds interact strongly → elute late.
Nonpolar compounds elute early.
Applications
Lipids
Fatty acids
Isomers with small polarity differences
Water-sensitive compounds
Ion exchange HPLC
Principle
Separation based on charge attraction between analytes and charged stationary groups.
Types
Cation-exchange (negatively charged stationary phase binds cations)
Anion-exchange (positively charged stationary phase binds anions)
Key Factors
Buffer pH (controls ionization of analytes)
Ionic strength (salt concentration controls elution)
Applications
Amino acids
Peptides
Nucleotides
Charged metabolites
Water-soluble vitamins
Protein charge variants
Size exclusion HPLC
Principle
Separation based purely on molecular size, not chemistry.
Large molecules cannot enter pores → elute first.
Small molecules enter pores and are delayed → elute later.
Applications
Proteins
Polysaccharides
Polymer molecular-weight distribution
Aggregation analysis (biopharmaceuticals)
HPLC: partioning principle
The fundamental mechanism is differential partitioning.
An analyte repeatedly moves:
Into the stationary phase
Back into the mobile phase
The fraction of time spent in each phase determines retention.
Partition Coefficient
Higher K → stronger retention → longer elution time.

Adsorption vs partition chromatography
Adsorption Chromatography
Analytes adsorb to surface sites on the stationary phase.
Common in normal-phase HPLC.
Partition Chromatography
Analytes dissolve into a bonded stationary phase layer (e.g., C18).
Dominant in reverse-phase HPLC.
HPLC: selectivity

Retention factor

HPLC: retention factor

Effects of Mobile-Phase Composition
In Reverse-Phase HPLC
Higher organic % (more ACN or MeOH) → shorter retention.
Higher water % → longer retention.
In Normal-Phase HPLC
Higher polarity solvent → shorter retention.
Lower polarity solvent → longer retention.
Role of pH in HPLC
For ionizable compounds, pH strongly influences retention.
If analyte is acidic:
Low pH keeps it neutral → more retention in RP-HPLC.
High pH ionizes it → less retention.
If analyte is basic:
High pH keeps it neutral → more retention.
Low pH protonates it → less retention.
pH also affects stationary-phase stability.
Temperature in HPLC
Increasing temperature:
Lowers mobile-phase viscosity
Sharpens peaks
Reduces retention
Improves mass transfer
Temperature must be controlled for reproducibility.
HPLC solvent properties
Polarity
Controls retention strength.
In reverse-phase (RP), increasing organic solvent (ACN/MeOH) decreases retention.
In normal-phase (NP), increasing polarity of solvent decreases retention.
Viscosity
Influences column pressure.
Higher viscosity → higher backpressure.
Acetonitrile has lower viscosity than methanol.
UV Cutoff
Important for UV detection.
Solvents must not absorb strongly at the detection wavelength.
ACN and methanol have low UV cutoffs.
Miscibility
Solvents must mix uniformly.
Water–ACN–MeOH combinations are fully miscible.
Hexane (normal-phase) is immiscible with water.
Common HPLC solvents
Water (HPLC-grade)
Main component in RP-HPLC
Must be low in particulates, organic impurities, and ions
Acetonitrile (ACN)
Lower viscosity → lower pressure
Strong elution strength in RP
Low UV cutoff (~190 nm)
Methanol (MeOH)
Slightly higher viscosity than ACN
Different selectivity compared to ACN
UV cutoff ~205 nm
Buffers in HPLC
Common Buffers
Phosphate buffer (strongest buffering range, not MS-compatible)
Acetate buffer
Formate buffer (MS-compatible)
Ammonium acetate/formate (volatile; ideal for LC-MS)
Important Buffer Guidelines
Keep buffer concentration between 5–50 mM.
Filters (0.22 µm) are required to prevent damage to pumps and columns.
Avoid buffers with non-volatile salts in LC-MS.
Ensure buffer pH is at least ±1 unit from analyte pKa for pH stability.
HPLC: Additives in mobile phase
Acids
Formic acid (0.1–1%)
TFA (0.05–0.1%)
Acetic acid
Uses:
Control pH
Improve peak shape of basic molecules
Suppress ionization for UV detection
Bases
Triethylamine (TEA)
Ammonia
Uses:
Improve peak shape of acidic analytes
Reduce interactions with residual silanols
Ion-Pairing Agents
Tetrabutylammonium salts
Sodium dodecyl sulfate
Uses:
Allow separation of highly polar or ionic compounds
Form neutral complexes to increase retention in RP-HPLC
HPLC: Column structure
Typical Dimensions
Length: 50–250 mm
Internal diameter (ID): 2.1–4.6 mm (analytical)
Particle size: 1.7–5 µm (common), sub-2 µm for UHPLC
Materials
Stainless steel housing for high pressure.
PEEK or titanium used in bio-compatible systems.
End-fittings include frits that retain the packing material but allow solvent flow.
Purpose of prep HPLC
Preparative HPLC is used when:
You need a pure chemical for further experiments
You need to isolate natural products from plant extracts
You want to separate and collect drug intermediates
You need to purify proteins, peptides, or metabolites
You want to produce reference standards
It focuses on yield, purity, and recovery, not analytical resolution.
Diff between prep and analytical HPLC

Key components of HPLC
Large-Diameter Columns
Internal diameter of 10–50 mm or even larger
Packed with larger particles (10–25 µm) to reduce backpressure
Designed for durability and large sample loads
High-Capacity Pumps
Deliver 10–100+ mL/min
Must handle high solvent consumption and heavy loads
Fraction Collector
Automatically collects eluted peaks into tubes/bottles
Triggered by detector signal or retention time
UV Detector
Monitors when target compound is eluting
Works at higher absorbance ranges due to larger sample loads
How prep HPLC works
Crude sample is dissolved in a compatible solvent.
Sample is injected in a relatively large volume.
Under high-flow, analytes separate on the column.
The target peak is detected with UV.
A fraction collector captures the desired peak.
Solvent is removed (rotary evaporator/freeze dryer).
You obtain pure isolated compound.
The purified compound can then be used for:
Biological assays
Structural characterization (NMR, MS, IR)
Drug discovery
Synthetic chemistry steps
Scaling from analytical to prep HPLC

Advatnages of prep HPLC
Produces high-purity compounds
Handles complex mixtures
Scalable from mg to gram quantities
Highly reproducible
Limitations of prep HPLC
Expensive (solvents, large columns, high-flow pumps)
Requires careful method optimization
Solvent waste is very high
Peak purity sometimes lower than analytical HPLC (must re-purify)
Gas chromatography principle
1. Basic Principle
The sample is vaporized in a heated inlet.
An inert carrier gas (e.g., helium, nitrogen, hydrogen) carries the vaporized sample through a long capillary column.
Compounds interact differently with the stationary phase lining the column.
More volatile compounds travel faster; less volatile ones spend more time interacting with the column.
A detector measures compounds as they elute, producing a chromatogram.
Components of gas chromatography (carrier gas supply, injector, GC column)
2.1 Carrier Gas Supply
Helium, nitrogen, or hydrogen
Must be pure and moisture-free
2.2 Injector / Inlet
Heats sample to instant vaporization (typically 200–300°C)
Split or splitless injection modes control sample amount
2.3 GC Column
Most GC systems use capillary columns:
Long (30–60 m)
Very narrow (0.2–0.32 mm ID)
Coated internally with stationary phase (e.g., nonpolar PDMS, polar PEG)
Column is placed inside a temperature-controlled oven.
Components of gas chromatography (Oven, FID)
2.4 Oven
Precisely controls temperature
Can run isothermal or temperature programs
Temperature programming improves separation of compounds with different boiling points
2.5 Detector
Common detectors include:
Flame Ionization Detector (FID)
Thermal Conductivity Detector (TCD)
Electron-Capture Detector (ECD)
Mass Spectrometer (GC-MS)
How separation occurs in gas chromatography
Two factors determine retention:
3.1 Volatility
More volatile → lower retention time
Less volatile → higher retention time
3.2 Interaction with Stationary Phase
Nonpolar compounds retained on nonpolar columns by dispersion forces
Polar compounds retained on polar columns through dipole interactions
Column chemistry determines selectivity
Gas chromatography detectors
FID (Flame Ionization Detector)
Most common
Very sensitive for organic compounds
Measures ions produced when compounds burn in a hydrogen flame
TCD (Thermal Conductivity Detector)
Universal detector
Measures change in thermal conductivity of carrier gas
ECD (Electron Capture Detector)
Sensitive to halogens, nitriles, pesticides
Common in environmental analysis
GC-MS
Most powerful
Provides both separation (GC) and identification (MS)
Widely used in forensics, toxicology, and metabolite identification
Applications of gas chromatography
Environmental pollutant analysis
Fragrance and essential oil profiling
Petrochemical analysis
Forensic drug testing
Food and flavor chemistry
Advantages of GC
Extremely high resolution
Fast analysis
Highly sensitive
Excellent for volatile compounds
Often coupled with MS for identification
Limitations of GC
Cannot analyze non-volatile or thermally unstable compounds
Derivatization sometimes required to make samples volatile
Requires dry, clean samples
GC-MS steps
2. How GC-MS Works (Step-by-Step) Step 1 — Injection
Sample is injected into the heated inlet (200–300°C), instantly vaporizing it.
Step 2 — Separation (GC part)
An inert carrier gas (helium, hydrogen, nitrogen) pushes vapors into a long capillary column.
Compounds separate based on:
Boiling point
Polarity
Interaction with stationary phase
Each compound exits the column at a different retention time.
Step 3 — Ionization (MS part)
Eluting molecules enter the mass spectrometer ion source.
Most systems use Electron Ionization (EI):
High-energy electrons (70 eV) strike molecules
A positively charged molecular ion forms
Molecules fragment into characteristic pieces
Step 4 — Mass Analysis
The fragments move through a mass analyzer.
Common analyzers:
Quadrupole
Time-of-flight (TOF)
Ion trap
Orbitrap (rare in GC-MS)
Fragments separate based on mass-to-charge ratio (m/z).
Step 5 — Detection
A detector (electron multiplier) amplifies ion signal and sends it to data system.
Step 6 — Mass Spectrum
Each compound produces a unique spectrum showing:
Molecular ion peak (M⁺)
Fragment peaks
Base peak (most intense peak)
This acts like a “fingerprint” for identification.
GC-MS outputs
You get two types of data:
Chromatogram
Plots signal vs time
Peaks = separated compounds
Retention times help narrow down identity
Mass spectrum
Plots ion abundance vs m/z
Used for exact identification
Compared to spectral libraries (e.g., NIST database)
GC-MS essentially gives both separation and structural identity.
GC-MS ionization method
4.1 Electron Ionization (EI) — Most Common
70 eV electrons
Produces reproducible fragmentation
Generates rich spectra
Rough on fragile molecules (hard ionization)
4.2 Chemical Ionization (CI)
Softer method
Produces more intact molecular ions
Better for determining molecular weight
Core idea of TLC
TLC separates components of a mixture based on differences in polarity and their affinity for a stationary phase vs. a mobile phase.
It is a quick, inexpensive, and qualitative technique used widely in biochemistry, organic chemistry, and analytical labs.
TLC materials
Stationary phase: A thin layer of silica gel (SiO₂) or alumina coated on a plate (usually glass, plastic, or aluminum).
Silica is polar and can form hydrogen bonds.Mobile phase: A solvent or solvent mixture (e.g., hexane:ethyl acetate).
The solvent can be non-polar, polar, or mixed, depending on what you want to separate.Sample: Small drop of your dissolved mixture.
Developing chamber: A jar with solvent at the bottom and a lid to create a saturated atmosphere.
Mechanism of TLC
TLC relies on partitioning:
Molecules that interact strongly with the stationary phase (more polar) move slowly.
Molecules that interact weakly with the stationary phase (less polar) move quickly with the solvent front.
Balance of:
Adsorption onto stationary phase
Solubility in mobile phase
Because your mixture’s components have different polarity, they move at different speeds → distinct spots appear.
TLC procedure
Draw a baseline in pencil (ink would dissolve).
Spot your sample on the baseline using a capillary tube.
Place plate in chamber with solvent; ensure the baseline stays above solvent level.
Solvent rises by capillary action and carries compounds.
Remove plate when solvent is near the top.
Mark solvent front in pencil immediately.
Visualize spots:
Under UV light (for UV-active compounds)
Using iodine chamber
Spray reagents (ninhydrin for amino acids, anisaldehyde, etc.)
Rf in TLC

TLC applications
Checking purity of a compound
Monitoring reaction progress (e.g., reactant disappears, product appears)
Identifying compounds by comparing Rf values with known standards
Selecting a good solvent system for column chromatography or preparative TLC
Distinguishing isomers or closely related molecules
polarity in TLC
Stationary phase (silica): very polar
Mobile phase: you choose polarity
Solvent choice determines movement:
If solvent is too polar → all spots go up too far (Rf too high)
If solvent is too non-polar → nothing moves (Rf ~0)
Best solvent: gives good separation (spots spaced out).
Advantages of TLC
Fast (few minutes)
Uses little solvent
Cheap plates and equipment
Can run many samples side-by-side
Easy visualization
Limitations of TLC
Mostly qualitative, not quantitative
Low resolution compared to HPLC
Not automated
Not good for volatile samples
Hard to scale beyond preparative TLC