Breast Cancer PY367
1/249
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
250 Terms
What are the risk factors for breast cancer?
60 years old
Oestrogen exposure: HRT, oral contraception, early/late menopause
23% Linked to lifestyle factors like:
Overweight
Alcohol
Certain occupational exposures
Family history
Inheritance of mutated BRCA gene (5% of cases)
Breastfeeding and physical activity protect against breast cancer
What is the presentation of BC?
Lump in breast
Change in shape or size
Dimpling of skin/thickening of tissue
Inversion of nipple
Rash on nipple
Discharge from nipple
Swelling or lump in armpit
What 2 symptoms are associated with BC?
Abnormalities of overlying skin
Unilateral bloody nipple discharge
How is it screened?
Breast self-exam
Has high false positive and negative rates
Not decreased mortality
Mammography
UK screening programme screens all women aged 50-70 every 3 years
Detection of early BC reducing mortality by 20-30%
Treatment Options
Endocrine therapy
Radiotherapy
Chemotherapy
MAbs and other targeted treatments
Patient pathway from appointment onwards
GP app. for suspicious lump
Physical exam, Radio, core biopsy, FBC, LFT, bone profile
Suspicion of metastasis leads to bone scan and chest abdo pelvis (CAP) CT scan
Immunohistochemistry - ER/PR/HER2
FISH (fluorescence in-situ hybridisation) part of HER2 testing using DNA primers linked to fluorescent marker
Detecting antigens in cells of tissue exploiting principle of Ab binding to them
Intensity of staining scores expression levels
BC MDM
Surgery
MDM post-surgery pathology - gene testing of tumour
Chemotherapy
What is TNM staging?
Tumour size
Lymph nodes
Metastasis
Used for all solid tumour growth and most common for BC
Explain T
T1 = under 2cm
T2 = 2-5
T3 = over 5
T4 = extension to chest wall or skin
Explain N
N1 = Mobile ipsilateral lymph nodes
N2 = fixed to one another or other structures
N3 = Intraclavicular or ipsilateral internal mammary and axillary nodes
Explain M
M0 = No distant metastases
M1 = Contralateral lymph nodes or any distant metastases
MX = Distant that cannot be assessed
Explain the numerical staging system
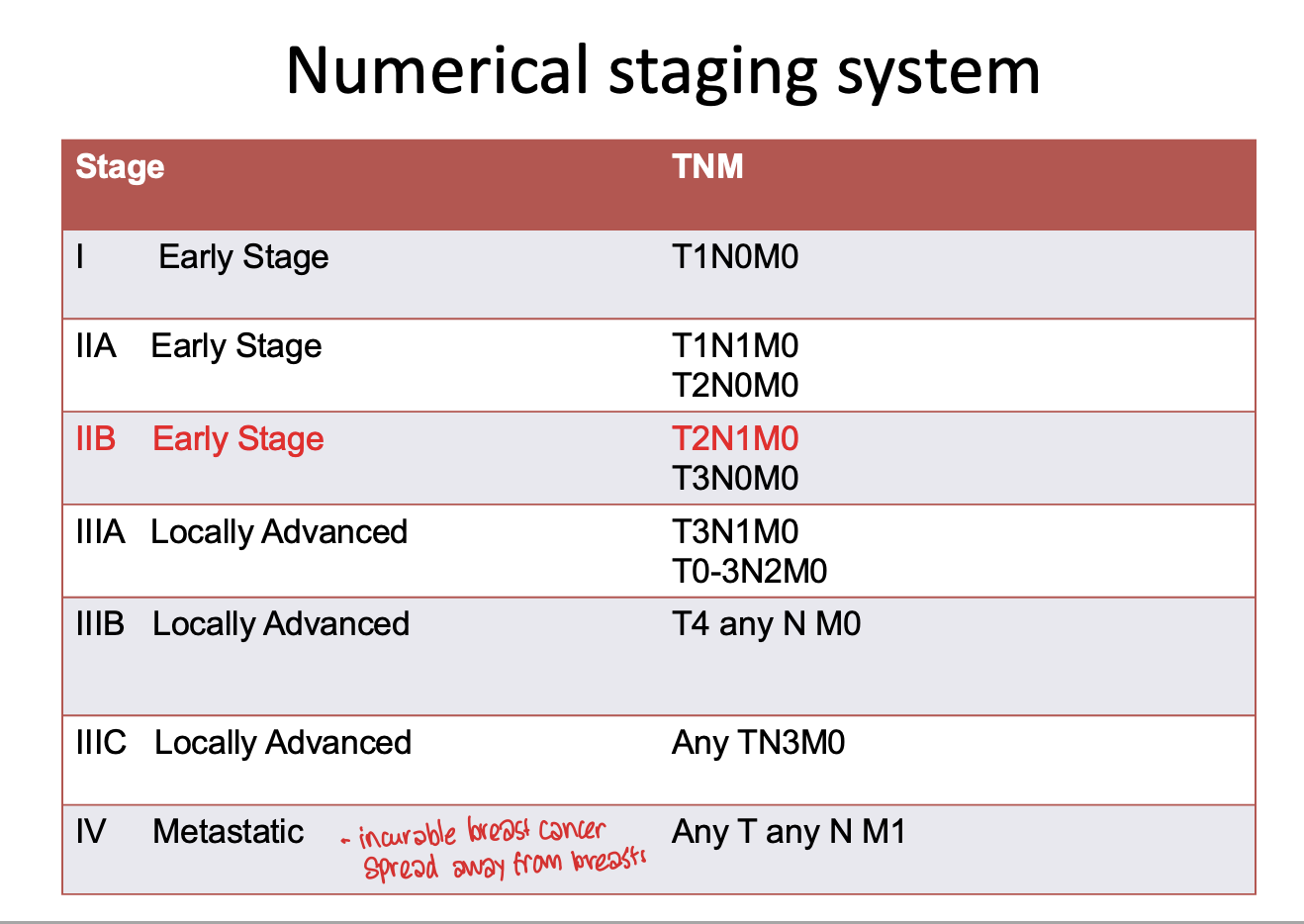
What is the grading pathology?
Grade 1
Well differentiated/low grade
Cancer cells similar to normal cells and grow slowly
Grade 2
Moderately differentiated
Look abnormal and slightly faster growing
Grade 3
Poorly differentiated
Look very different and grow fast
Pathological Classification
Ductal 70-80%
Worst and most invasive
Milk ducts
Lobular 5-10%
Bilateral (lobules = glands that make the milk in outer breast)
Lesions in same area
Tubular 10-20%
Medullary 5-10%
Good prognosis because it grows slower than ductal and doesn’t usualy spread outside of breasts (i.e. lymph nodes) therefore easier to treat
Invasive - starts in milk ducts and then spreads into surrounding tissue
Mucinous/colloid 1-2%
Other 1-2%
Inflamm 2%
50% survival at 5 yrs due to blockage of lymphatic drainage = reason why we see peau d’orange
Immunohistochemistry
ER and PR - Hormone dependent tumour
Favourable diagnosis
More likely to respond to treatments e.g. Tamoxifen
HER2 positive - Transmembrane tyrosine kinase which regulates growth, survival and migration
More aggressive and less favourable
Responds to Trastuzamab
What is hormone receptor negative?
No target to inhibit tumour growth (10-15%)
Triple negative BC and more common in younger women, afro, BRCA mutation
HER2+ tumours dependent on female hormones for growth and survival
What is luminal A?
One of most common
Start in the inner cells lining mammary ducts
Tend to be ER+ and/or PR+ and HER2 -ve and tumour grade 1/2
Less than 15% have p53 mutations = best prognosis and high survival rates
ER+, treatement usually involves hormonal therapy
Explain Luminal B
ER+ and/or PR+
Tend to have mitotically active cells, positive for Ki67
Often HER2+
Diagnosed at younger age than luminal A
Poorer prognosis with poorer TNM
Triple negative/basal like
ER-, PR- HER2-
Subsects like basal-like
Tumour have cells with similar features to basal cells
Most BCA1 BC are both triple negative and basal like
Younger women, Afro-american women and BRCA mutation
Often aggressive and have poorer prognosis
Treated with combo of surgery, radiation and chemo
HER2 Type
Not same as HER2+ and not used to guide treatment
These tend to be ER and PR negative with lymph node involvement and poor tumour grade
Fairly poor prognosis and prone to early and frequent reoccurrence and metastases
People tend to be diagnosed at a younger age than those with luminal
HER2 tumours can be treated with Trastuzumab
What is claudin low
Often - - - but distinct in there is low expression of cell-cell junction proteins including E-cadherin and there is frequent infiltration of lymphocytes
Also enriched in mesenchymal and stem cell features
Explain Normal like
Usually small and tend to have good prognosis
How to lower risk of reoccurrence after surgery?
Adjuvant Chemo
Name a molecular assay and how is it used?
Oncotype DX
Real time PCR on 21 genes causing tumour proliferation
Validated reoccurrence score indicating 10 year risk
Validated prediction as to whether patient will have additional benefit from chemo compared to Tamoxifen alone
Usually luminal A pts.
What is neo adjuvant chemo?
Starts before surgery to shrink tumour and improve outcomes
Used in:
Locally advanced tumours
Inflam tumours
Preserve tissue and facilitate less invasive surgery
What is used in adjuvant chemo HER2+ve?
Cyclophosphamide
Anthracycline
Taxane
What are Cyclophosphamides?
Alkylating agent - crosslinks DNA strands and inhibit DNA synthesis, transcription and replication
Alkyl groups to guanine base
Interferes with DNA replication by forming intrastrand and interstrand DNA crosslinks
Non-cell cycle specific so works regardless of what part the cycle is in
Pro-drug - activated in liver
What are anthracyclines?
Create free radicals = Ox damage
Intercalates between base pairs in DNA
Inhibit action of topoisomerase 2 by stabilising DNA-topoisomerase 2 complex and preventing re-ligation of double helix
Non-cell cycle specific
Can cause red urine
What are Taxanes?
Enhance polymerisation of tubulin
Stabilise microtubule polymer preventing disassembly of mitotic spindle
Blocks progression of mitosis resulting in apoptosis
M phase of cell cycle
Stops spindle from separating into two cells because cell is frozen in M phase = cell death
How is combo chemo used?
Drugs act at different cell cycle stages to maximised cytotoxic effect
FEC-T regime
5-Fluorouracil
Epirubicin
Cyclophosphamide
Docetaxel
5-F could be removed without affecting efficacy due to new evidence but not adopted in practise yet
MOA of Trastuzumab
Tags cell to IS
Blocks receptor dimerisation with other receptors
Stops signalling downstream into cell preventing proliferation
Issue with Trastuzumab?
Too expensive (25k) per pt.
Available as Biosimilar now though
Saving millions and opening up access
What is used with Trastuzumab?
Pertuzumab and trastuzumab combination provides a more comprehensive block
When used together they can block both sides of the HER2 receptor effectively to prevent dimerization as opposed to Herceptin alone
What can we learn about studying cell cycle control?
Development
Stem cells
Differentiation - occurs whether cells get the right cell
Cloning and processes of differentiation for cloning tissues or whole organisms
Division capacity - after division for a certain amount of times, cells go into senescence
Senescence
Oxidative damage effects
Opportunities for therapeutic intervention
Stopping cell division that leads to carcinogenesis
3 ways of studying cell cycle
In whole organisms e.g. yeast
In cell-free extracts e.g. frog eggs
In cell culture e.g. mammal cells
Phases of the cell cycle
G1 - 1st gap phase
Cells increase in size
Production of RNA and protein
Damage check
S - synthesis phase
Takes 6-8 hours
All DNA replicated
Replication bubbles and forks in different directions
G2 - 2nd gap phase
2-6 hours
Cell growth - protein production and sorting of organelles
Check for replication errors
M - Mitosis phase
Shortest phase
Compartmentalisation
Chromosomes are partitioned into two daughter cells
Why do we use yeast models to look in between phases?
Rapid production
Genome size
Amenable to genetic manipulation so we can easily change to track part of cells
Gene deletions, replacement or alteration
Can use fluorescence to track better
Can proliferate in haploid state
Can make temp sensitive mutants which stop functioning at certain temperatures
Helps understand cyclin dependent kinases and cyclins
Pauses the replication cycle at a certain stage so we can see what involved within that stage (e.g. pause in G1 and then see what is involved in that stage)
3 ways of identifying cell cycle stages
Radiolabelled nucleotides and X-ray photography
Cells take these up at replication so they’re more easily visible to see stages
Artificial analogues and Ab staining
Modded by adding fluorescent ab which allows tracking of cell division
Flow cytometry
Stain all DNA with fluorescence indicating the phase
Cells in G1 phase contain half the DNA of cells after G2 and M
Cells in S phase contain intermediate quantity
What 2 major points are cell cycle events controlled at?
Entry point to cell cycle
Critical checkpoints
What choices does a cell have when its at division?
Divide
Not divide
Apoptosis
Quiescence - Cell steps out and goes to G0 phase and has ability to return to cell cycle when environment is right
Senescence
Entry into cell cycle due to growth factor availability
Restriction point at late G1 due to growth factor availability
If no growth factors, cell enter quiescent stage G0
If cell passes restriction point = division irreversible
Cells normally have limited proliferation capacity = enter G0 permanently called senescence
If cycles are disturbed or damaged they will not enter replicative senescence = IMMORTALITY
What happens at each checkpoint?
G1 detects presence of damaged DNA and leads to cell cycle arrest
Checks if DNA intact and nothing wrong
Cascades activated if damage to CKIs, tumour suppressor proteins
G2 checkpoint arrests cells in response to damaged or unreplicated DNA
Depending on extent of damage, cell goes into senescence or apoptise or repairs
Ensures all DNA replicated, if parts missing = improper proliferation
M phase checkpoint arrests mitosis if the daughter chromosomes are not properly aligned on mitotic spindle
Misaligned = pause cycle
If they cannot realign, cell does not continue - important in cancer targeting
What is cell cycle progression determined by?
Cyclin dependent kinases
These are protein kinases whose activity will rise and fall during cell cycle
Phosphorylation of intracellular proteins/enzymes initiate or regulate major events of cell cycle
This going wrong causes issues
CDKs partially activated by cyclins and regulated by multiple processes
What happens to cyclin levels over time?
Increase during interphase and decrease during mitosis
Accumulate during interphase and rapidly degrade towards end of mitosis
What is the process of removal of cyclins?
After phosphorylation, cyclins are degraded by ubiquitin/protease system
Ubiquitin tags cyclins after phosphorylation event
Why is the availability of them key to cell cycle control?
Many different types of cyclins with different roles
Controls activity of CDKs and promotes cell progression
When CDKs are complexed with appropriate M-phase cyclin, mitosis machinery is triggered
When CDKs are complexed with appropriate S-phase cyclin, trigger DNA replication
Cyclin sub-groups and CDKs
Cyclin A = CDK 1/2
B = 1
D = 4/6
E = 2
Precise timing of each step is essential and many cycle proteins are degraded after they have carried out their functions
How is activity of these complexes negatively modulated at either of the checkpoints?
In G0, Cyc D is present at low concentration and Rb protein is hypophosphorylated restraining cell at checkpoint 1 by inhibition of several proteins which are critical for cycle progression
What is the Rb protein and what does it do?
Tumour suppressor protein
Binds transcription factors and prevents them from promoting expression of genes required for DNA replication during S-phase
Each molecule present during different phases of cell stages
CDK1 only one required to drive through cell stages
Cyclin B1 and A2 essential for normal cell cycle
Phase | CDK | Cyclin | Inhibitor |
G1 | CDK 4, 6 | Cyclin D | P16, P21, P27 & P15 |
G1/S | CDK 2 | Cyclin E | P21, P27 |
S | CDK 1, 2 | Cyclin A | P21, P27 |
M/G2 | CDK 1 | Cyclin B | (none) |
What is the mechanism of CDK regulation?
Association of CDK with its cyclin
Phosphorylation events
Phosphate attached to threonine amino acid
Causes repellent force on amino acid causing conformational change in their structure allowing for inhibition (thr14+tyr15) or activation (thr160)
Association with CKI stops action of CDK/CYC complex
Positive CDK regulation by phosphorylation
Without cyclin, CDK remains inactive therefore by moving T-loop, active site opens allowing cyclin to bind
Once CAK phosphorylates Thr160, T-loop is fully removed which opens active site
Negative CDK regulation by phosphorylation
If complex is made early in cell, needs to remain inactive till required
WEE1 provides inhibitory phosphorylation
CDC25 dephosphorylates afterwards to allow for cell progression
Processes determine wether WEE1 or CDC25 is up or down regulated
How do CKIs regulate CDKs and two family types?
They bind to complex and distort active side by inserting into ATP binding site
Ink4 family - P15, 16, 18, 19 Inhibit CDK4/6 = entry into cell
Cip/Kip family - P21, 27, 57 Inhibit CDK1/2 = whole cycle
P21 and 16 dominant inhibitors of cell proliferation in senescent cells
Control of cell cycle entry
Growth factors regulate cycle progression through G1 restriction point
Cyclin D via MAPK signal path
IF NOT CONTROLLED = UNCONTROLLED CELL DIVISION = CANCER
CDK4/6 with cyclin D drive the passage through restriction point
Receptor protein key area dysregulated in cancer
What is the cell signalling pathway?
Signal binds to receptor on RTK
Tyrosine kinase dimerise and autophosphorylate forming hyperphosphorylated tyrosine kinase complex
Adapter protein activated signalling and activating Ras activating protein
This happens indirectly from conversion of GDP to GTP
Activated Ras phosphorylates other molecules and starts intracellular cascade of signals
What does Ras now do?
Feeds into intracellular signal pathways e.g MAPK
Acting as phosphorylation cascade
Eventually alters activity of target proteins causing gene expression changes
Rafs → MEKs → ERKs
How does this initiate cell division?
MAPK cascade leads to transcription factors expression
Myc promotes transcription of cyc D → activates CDK4/6
G1 complex made and activated due to initial growth factor
What does the CDK4/6 complex do now regarding the Rb protein?
Rb usually binds and inactivates E2F proteins and is responsible for G1 restriction point
When cell is ready to divide at G1 restriction point, G1-CDK complex phosphorylates pRb-E2F complex to free E2F to act as transcription factor
CDK2/cycE complex also hyperphosphorylates pRb in G1
E2F acts on S-phase gene transcription making cyc E and A leading to cell cycle entry and DNA synthesis as cell can now go from G1 to S-phase
pRb protein now hyperphosphorylated and therefore inactivated
How does positive feedback loop ensure process continues when required?
Hyperphosphorylation of pRb is required to free E2F because it is a key transcription factor
Blocking CDK is not enough to block cell cycle as pRb can be phosphorylated by other pathways
E2F activates genes like PCNA and cyclins
Why is there DNA replication once per cycle?
Do not want extra parts which have more copies than others
Cannot get re-replication
Cannot start more cycles whilst one is running
Cycles can only go in linear forward direction
What do cancer chemo agents do?
Stop division by blocking DNA replication/damage DNA/block mitosis
Causes growth arrest by triggering cell cycle checkpoints with damage and errors
They also act via growth factor signal inhibition and reduce cell entry e.g. Herceptin
7 chemo agents and their actions
Methotrexate - Prevent folate synthesis for purine and pyramidines
Etoposide - Topoiso 2 inhibitors prevents religation of DNA
Irinotecan - As above for Topoiso 1
Bleomycin - Strand scission
Doxorubicin - Intercalator which prevents Topoiso 2 action and alters membrane fluidity
Emtansine - Trastuzumab helps enter cell and broken down to release mertanisine
Paclitaxel - Stabilise microtubules so no disassembly for elsewhere use
What is the human epidermal growth factors receptor?
Works same way as cell signal pathway above
When growth factor stimulates HER2 ligands, TKase domains dimerise and hyperphosphorylate which trigger same Ras/Raf/Mek/Erk early response pathway to drive cell cycle
What is the alternate P13k pathway?
Goes through AKT which is linked to apoptosis inhibition and mTOR which is linked to protein biosynthesis
Also blocks CKIs
How does herceptin work?
Many HER2 receptors proliferate causing too much cell growth
Herceptin blocks these to stop signal
Cyclin timing
Decision to replicate requires E2F initiation by growth signals
These stimulate CDK activity by antagonising CKI and expressing cyc D
Rb inactivates E2F
Commitment to replication requires cyc A
Metaphase decision - replication stress checkpoint
What happens at G1 /DNA damage checkpoint?
G1 checkpoint mediated by P53
P53 levels increase in response to levels of DNA damage
Cancers occur to P53 mutations as they bypass checkpoint
What does DNA damage lead to an increase of?
Increase of P53 which increases P21 levels
This blocks machinery driving restriction point
What is P21?
Effector
Acts on G1, G1/S and S-phase CDKs
Can be activated independently (without P53)
Can change regulation cyc D activity
What are the roles of P53?
Causes transcription of P21 which is also a CDK inhibitor
CDK inhibition via P21 prevents cell cycle progression
P21 can also bind and inhibit PCNA
PCNA is a component of DNA replication machine that prevents it
It can also stop cell division to allow for DNA repair
If irreparable, it initiates signalling pathway which causes cell apoptosis
Apoptosis process
P53 transcriptionally activate pro-apoptotic genes
Bax
Fas
Bcl-2 promotes survival by inhibiting apoptosis factors but is downregulated when a cell needs to be apoptosed
This can go wrong in cancer
Bcl-2 upregulated and Bax downregulated
What are the 2 routes of cell death?
Necrosis
Cells spill contents into surrounding tissue and evoke an inflamm response
Apoptosis
DNA is systemically fragmented
Cell contents are packed into membrane vesicles and phagocytosed by adjacent cells
How is G2 checkpoint controlled?
Mediated by chk1 kinase as well as P53
Chk1 inactivates cdc25 in response to unreplicated or damaged DNA
Without cdc25, CDK1/cycB will remain inactive and cells will arrest in G2
Some cancers end up over expressing
What is cdc25?
Phosphatase which dephosphorylates CDK1 in order to activate it
When DNA damage is repaired, inhibitory signal is turned off and cell cycle progression continues
How does cdc25 bypass G2?
Active cdc25 needs to dephosphorylate inhibitory phosphates on CDK1/cycB complex
What happens in S phase?
Prophase separates all the replicated material out
Disassembly of material and condensing of chromosome's
Start to see chromosomal material and get kinetochores and spindles aligning at the different poles of the cell
Microtubules come out and catch onto chromosomes at the centromere
Alignments at the checkpoint and then the cell goes through anaphase very quickly and reforms the envelopes and cytokinesis
How are levels of MPF/M-phase CDK complex controlled?
It is made much earlier and is activated by CAK160 but also deactivated by WEE1
Passing G2 starts cascade - Phosphorylating cdc25 phosphatase
cdc25 Dephosphorylates inhibitory phosphate groups on CDK1 making it active and traversing into M-phase
Positive feedback ensures full movement into M phase
Complex being active triggers degradation of cyclin
G2: Chk1 mediated phosphorylation inactivates cdc25
M-phase: CDK1 mediated phosphorylation activates cdc25
What happens if chromosome s aren’t attached during M phase?
Unattached kinetochores inhibit APC/C and therefore inhibit anaphase initiation
If chromosome is only attached at one side
This activates and recruits MAD2
RIT1 is a Ras-related GTPase which interacts with MAD2 to inhibit APC/C
CDK1 inhibits RIT1/MAD2 complex
Allows APC/C to initiate anaphase
Spindle tension is also detected by cell and if wrong, attachments are destabilised to try again
How is chromatid separation controlled?
Cohesion complex binds sister chromatids together
Cohesions need to be cleaved by separase before the chromatids separate in anaphase
Separase kept inactive by securin until degraded by proteolysis via APC/C
When cell is ready to proceed, M-CDK phosphorylates APC/C, facilitating cdc20 binding which activates complex
What happens when cdc20 binds APC
Complex releases inactive separase from securin by ubiquitinating securin
Once active, separase can now break down cohesins to allow movement from metaphase to anaphase
CDC20 triggers cyc B destruction
What 3 things happen when cell cycle control is lost?
Alterations in cell proliferation
Alterations in DNA damage response
Alterations in cell growth
2 things that lead to loss of control
p53 no longer acting as caretaker - no blocking of cell cycle
Rb can be permanently hyperphosphorylated and will release E2F to drive proliferation
What 2 genes are traditionally distinguished?
Oncogenes
Tumour suppressors
Explain Oncogenes
Overactive form of normal cellular genes
Example of proto-oncogene: Ras turns into an abnormal form that is permanently switched on so cell thinks it is permanently stimulated by growth factors
Can also enter cell as part of virus e.g. HPV - inserts oncogene into cells which causes predisposition to mutation
Oncogenes more associated with spontaneous, somatic cancers
Myc, Ras, C-Fos
Explain Tumour suppressors
Genes that usually inhibit cell proliferation and tumour development
In tumours, these are lost or inactivated
Requires two mutational events
What does oncogene being dominant mean?
Mutation only needs to occur in one of the chromosomes
What are 3 ways proto-oncogene can be made overactive = Oncogene?
Mutation in coding sequence - deletion or point mutation
Gene amplification- overproduce
Chromosome rearrangement
Cancer cells display abnormalities in chromosome structure in……
Translocations
Duplications
Deletions
What is the philadelphia chromosome (translocation)?
Reciprocal chromosomal translocation event
Found in most chronic myeloid leukaemia pts.
ABL1 gene from chr.9 and BCR gene from chr.22
ABL codes for TKase
BCR responsible for neutrophil function
Codes hybrid constitutively active TKase which results in uncontrolled division and genome instability through various signalling pathways
What are the function of proto-oncogenes?
Growth factors
Growth factor receptors
Elements of intracellular signalling pathways
Regulatory GTPases e.g. Ras
Cytoplasmic kinases e.g. Raf, CDKs
Anti-apoptosis factors e.g. Bcl-2
Transcription factors e.g. Myc
G:C to T:A mutational hotspots
Ras has a G to T transversion in codon 12 causing impaired GTPase function leaving Ras constantly on
Causes cell division irrespective of growth factor signalling
Why is it harder to lose tumour suppressor genes?
Recessive mutation
Associated with inherited cancers as pts. are born with one mutation and second is lost during lifetime
Because of this proliferation of cells always switched on
3 types of tumour suppressor genes
Gatekeepers: Monitor cell division and induce apoptosis e.g. Rb, CKI’s, apoptosis genes
Caretakers: Promote genome stability and oppose mutation rates via checkpoints e.g. P53, BRCA, MMR
Landscapers: Control cellular microenvironment e.g. cadherins, integrins
Prevents metastasis of cancer cells by keeping them connected with their environment
When mutated, contribute to neoplastic growth
Caretakers and the mutator phenotype hypothesis
Mutation occurs and inactivates DNA repair gene so the mutation can have further mutations
This eventually leads to proto-oncogene → oncogene
CANCER
What are BRCA 1 and 2?
Both caretaker tumour suppressor genes
What are BRCA mutations?
BRCA gene is tumour suppressor which creates protein to fix double stranded breaks in DNA from replication stress
Repairing them means cycle continues
BRCA mutations mean that the protein repairing DNA changes shape and is non-functional
Accurate Repair
Main pathway with best outcome
Uses homologous DNA template to fix DSB with exact nucleotides
Deletion pathways
Other pathways which aren’t as favourable but you can still repair if you’ve lost BRCA 2
Deletes large parts leading to chromosomal rearrangement but temporarily fixes DSB
Error prone repair
Loss of BRCA 1 and 2
Try join ends of DNA with what it can, uses incorrect nucleotides which can cause mutations
What are cytoskeletons?
Immediate filaments inside cells
Without which cell wouldn’t have shape and organisation or motility, contraction, tensile strength