Genetic Variation
1/33
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
34 Terms
Definitions
•A gene is a region of DNA that contains all of the information required to produce a functional protein or RNA
•A gene locus is the unique position of a gene on the chromosome. Each individual’s genome contains the same genes in the same order i.e. at the same loci
•Genome refers to all the genetic material (DNA) of an organism
•Genotype – the genetic constitution of either a genome or an individual locus. The genotype is distinct from its expressed features or phenotype.
•Phenotype – the expressed features of the genome and results from the interaction between the genotype and the environment
•Genetics – the study of single genes and their effects
•Genomics – the study of all the genes in the genome, how they interact and including their interactions with environmental factors
•Epigenetics the study of reversible, heritable mechanisms that influence gene expression without changing the DNA sequence.
•The wildtype is the most common form of the gene within a population
•An allele is an alternative form of a gene which has one or more differences in its nucleotide sequence.
•Some genes are biallelic (have two alleles), many have multiple alleles (polyallelic)
•Homozygous – when the copies of the gene (alleles) present on each of a pair of homologous chromosomes are identical
•Heterozygous – when the copies of the gene (alleles) present on each of a pair of homologous chromosomes are different
•If an allele is dominant its phenotype is displayed if one copy of the dominant allele is present
•If an allele is recessive its phenotype is not seen unless two recessive alleles for the gene are present
•An allele can also have incompletely dominance or be co-dominant.
Co-dominance: ABO Blood Groups
•Both alleles are expressed equally to similar effect - both alleles are expressed to the phenotype equally e.g. ABO blood group
•3 alleles for glycosyltransferase which is responsible for the type of oligosaccharides found on the RBC. There are 3 alleles A, B and O and 4 phenotypes A, B, AB and O.
•A: transferase A (N-acetylgalactosamine added)
•B: transferase B (D-galactose added)
•O: inactive transferase
•A & B are co-dominant, O is recessive as transferase is inactive and no final sugars are added to the precursor oligosaccharides that are present on the RBC surface. A deletion leads to a fame shift and therefore no activity and no sugars being added.
Genotype | Phenotype | Antigen | Antibodies |
AA | A | A | Anti-B |
AO | A | A | Anti-B |
BB | B | B | Anti-A |
BO | B | B | Anti-A |
AB | A B | A + B | none |
OO | O | - | Anti-A + Anti-B |
Genetic basis of the ABO blood groups

Incomplete dominance
•In the heterozygous both alleles at a gene locus are expressed, producing an intermediate phenotype where one allele has more effect than the other, but doesn.t completely mask the effect of the recessive allele
•
•
Homozygous for wildtype allele (TT)
full activity
Homozygous for variant allele (tt)
Low activity
•Heterozygous (Tt)
T = wildtype allele (full activity)
t = variant allele (low activity)
•intermediate activity (both alleles expressed and the dominant allele does not completely mask the recessive allele) e.g. hair texture - 2 alleles curly and straight, heterozygous alleles = wavy hair
Definitions
•A polymorphism is a difference in DNA that is observed at a frequency of equal or greater to 1% within a population
•Often have no visible clinical impact
•Unlikely to be the main cause of a disease
•A mutation is a difference in DNA that is associated with disease
•The haplotype is a set of DNA variations or group of alleles that are inherited together as they tend to stay together as a set
•Germline cells - those that contain and transmit genetic information from generation to generation - egg and sperm cells
•Somatic cells – all cells other than germline.
Sources of genetic DIFFERENCE
Offspring are genetically different
from their parents
from one another
•Independent assortment of homologous pair of chromosomes occurs in meiosis
•Random fertilisation also contribute to the differences
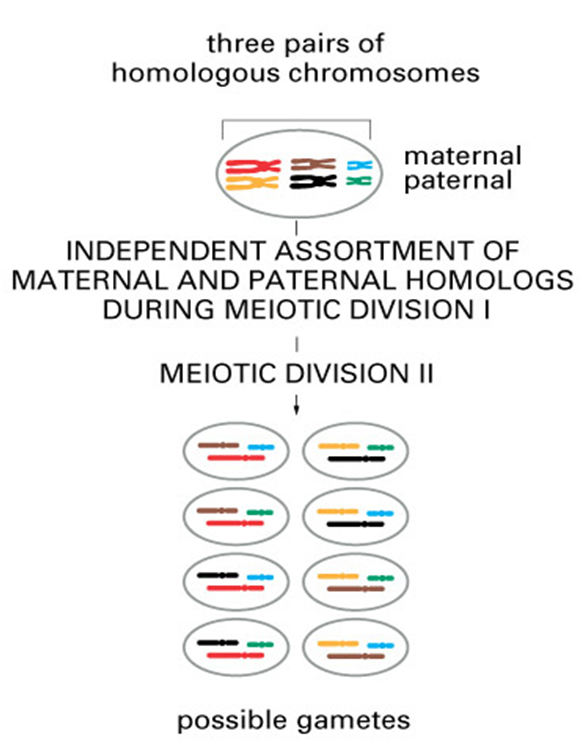
Sources of genetic DIFFERENCE
•Differences also arise due to crossing over - prophase 1. Homologous chromosomes which are composed of 2 sister chromatids some together and line up adjacent to each other. exchange genetic information by recombination, occurs during meiosis
•There are recombination “hot spots” which lead to the formation of blocks of sequence which are inherited as a single unit within a population – a haplotype block
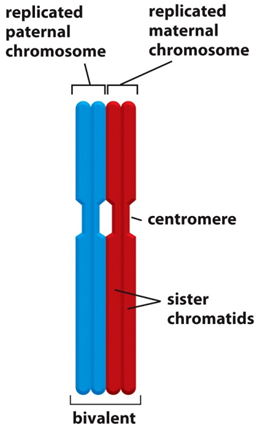
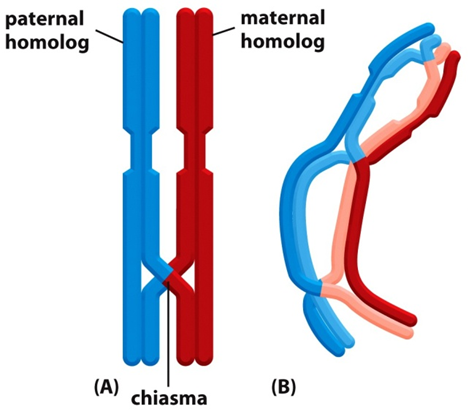
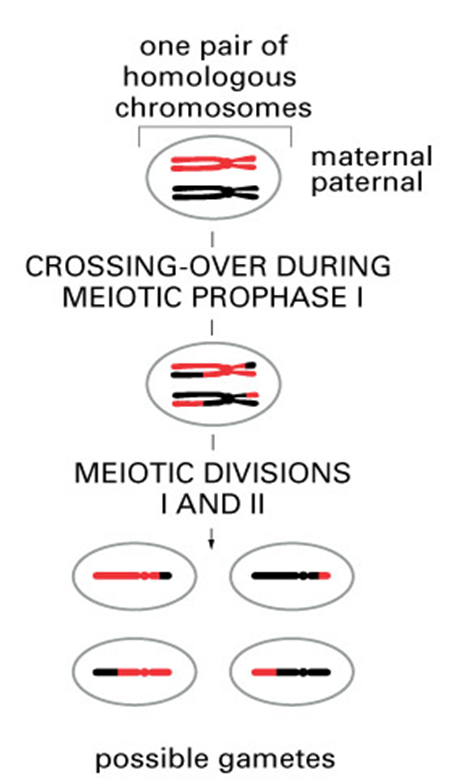
Sources of genetic variation
•Damage to chromosome ends can lead to their fusion and incorrect separation during meiosis causing gene amplification and loss
If a chromatid breaks one cells gains additional genetic material as a fused chromosome and the other has part of the chromosome missing
if the microtubules break then this results in one cell gaining both chromosomes and the other receiving neither
after DNA replication, loss of telomeres can lead to sister chromatids fusion. If chromatids break during cell division this leads to gene amplification in one cell and gene loss in the other.
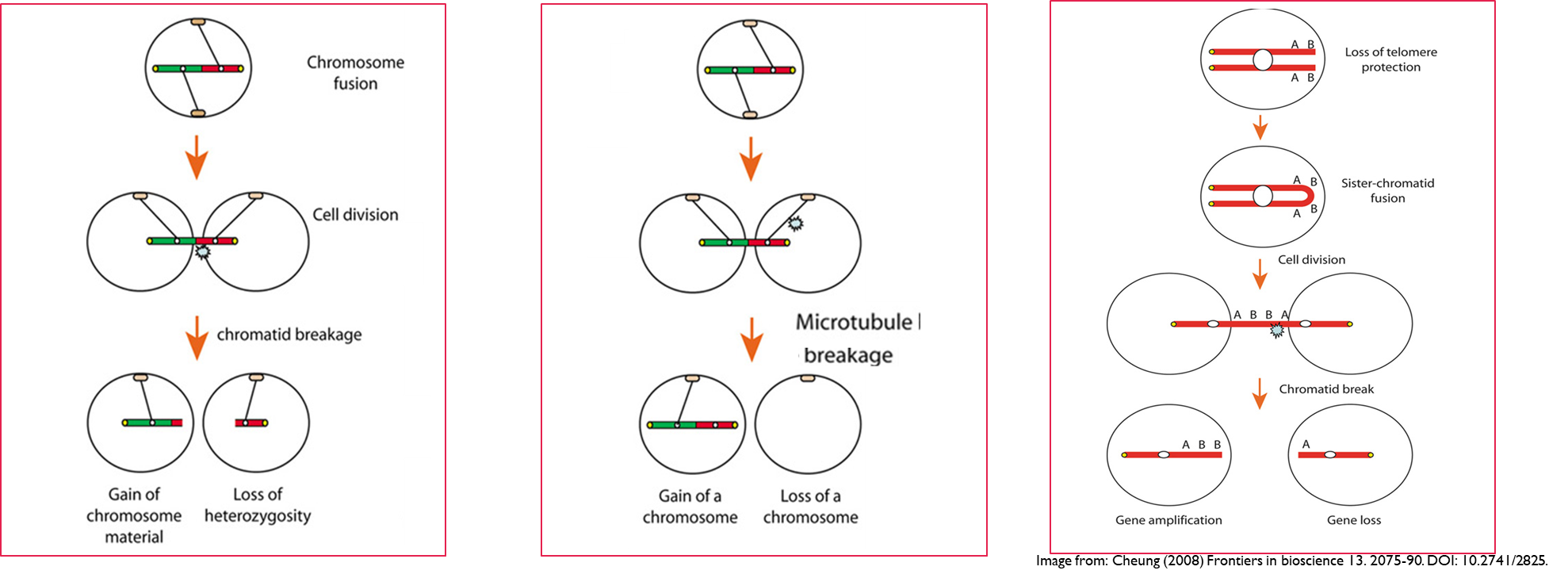
Sources of genetic variation
•Variations in DNA can be caused by:
•Errors in process of DNA replication
•Damage to DNA caused by environmental factors - environmental mutagens can attach to DNA and prevent the right base from being added
•These can be maintained in the germline DNA over time if they are
not “harmful” or
provide an advantage or
the variation does not have an impact on somatic cells until after reproduction.
Consequences of Genetic Variation
•For a variation to have a phenotypic effect it must occur in either the regulatory or coding region of a gene.
•If it occurs in the regulatory region it can either cause alterations in efficiency of:
•Transcription - variation has to occur in promoter region of the gene where transcription machinery binds and it recognises specific sequences within those binding sites. Cal lead to changes in levels of protein, therefore the gene will still be the wild type but there will either be more or less of it
•Translation - variation has to occur at the START sequence for translation - can also affect how much protein is produced.
•Splicing - interferes with splicing from pre mRNA to mRNA, has to happen before the mRNA leaves the nucleus for transcription
Consequences of Genetic Variation
•If it occurs in the coding region of a gene it can:
•Affect the amino acid sequence of the protein to alter its functional properties
•Have an effect on the rate of folding of the protein (a consequence of codon abundancy) - if a codon is changed to that of a much rarer transfer RNA this can slow down translation because it takes longer for this RNA to fit within the ribosome
•Have an effect on protein stability
Types of genetic variation
1.Single nucleotide polymorphisms
2.Base deletion or insertion
3.Microsatellites
4.Minisatellites
5.Copy number variations
Single Nucleotide Polymorphism
•Caused by base substitution
•Occur in the genome ~ 1 every 1000 bp
•Human genome is ~3.1 billion bp
•~ 3 million SNPs in an individuals genome
v
There are different types of SNP
Synonymous
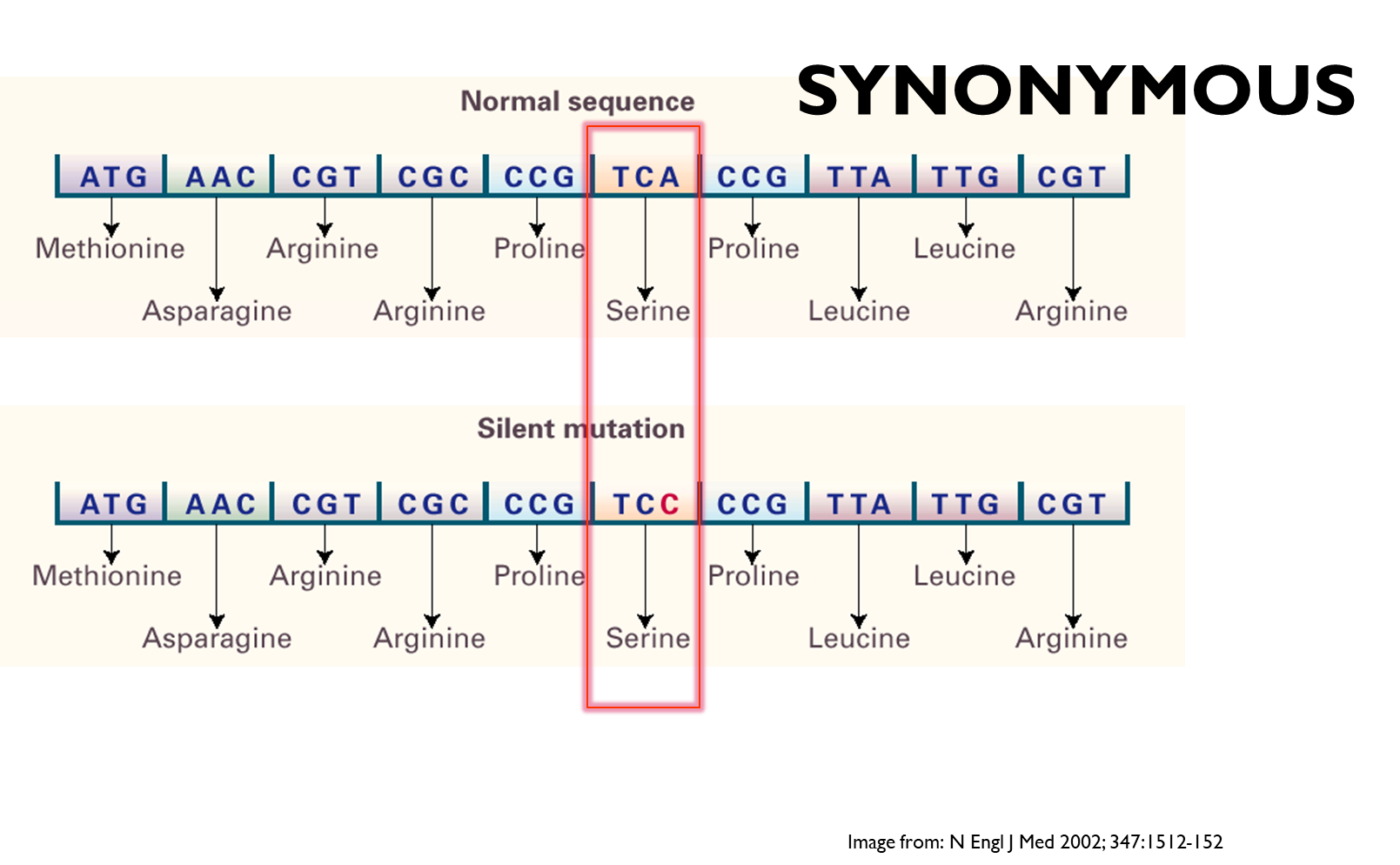
This is a silent SNP as although the codon change due to the different base it still codes for the same amino acid - no effect on the protein itself, but can have an effect on the rate of production of the protein if tRNA is changed to a more rare one
Non Synonymous
- Conservative missense
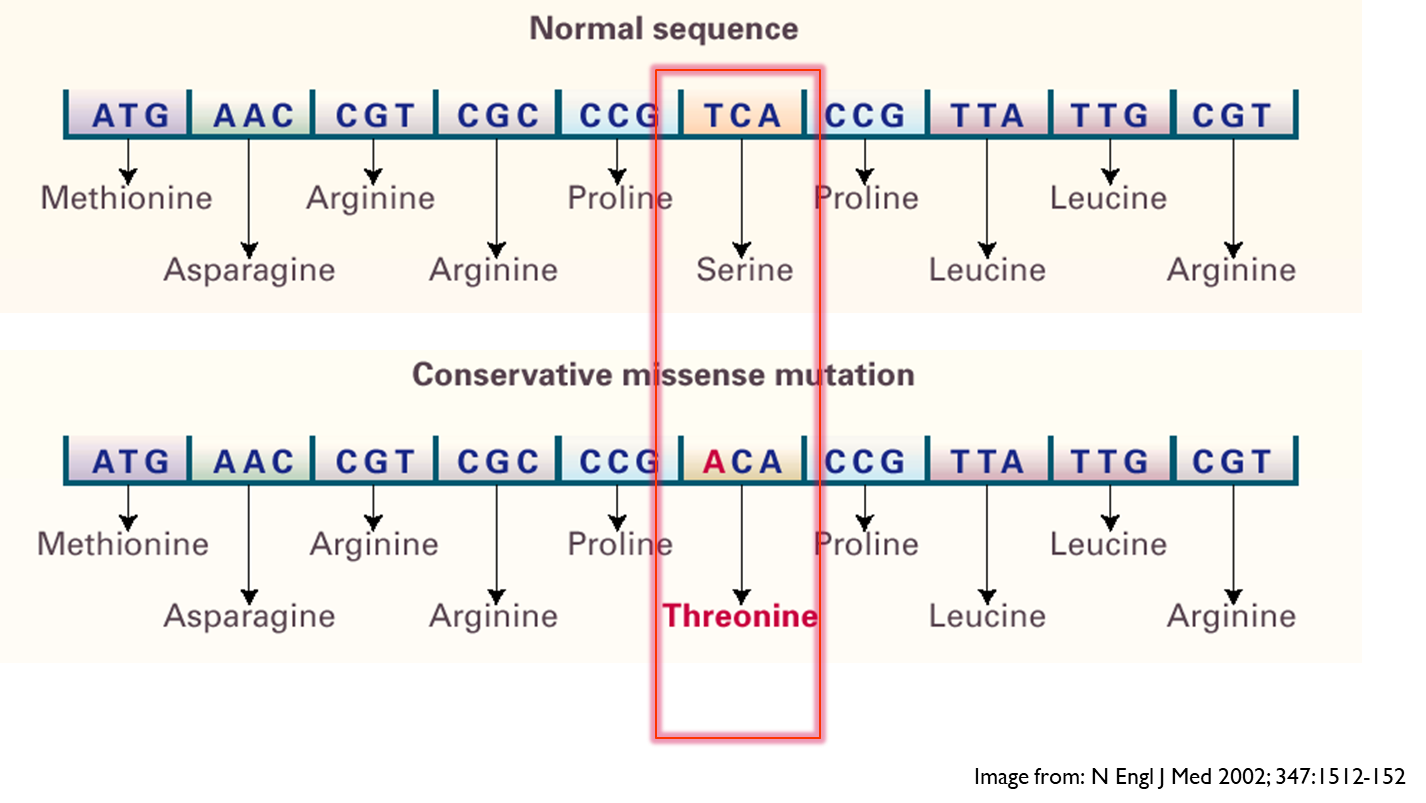
This is where the change in the nucleotide causes a change in the amino acid.
Different categories of this
Conservative missense change - here although the amino acid changed due to change in base, the alteration might only have a limited effect on the proteins function as both amino acids have uncharged and hydrophilic side chain
Non Synonymous
- Nonconservative missense
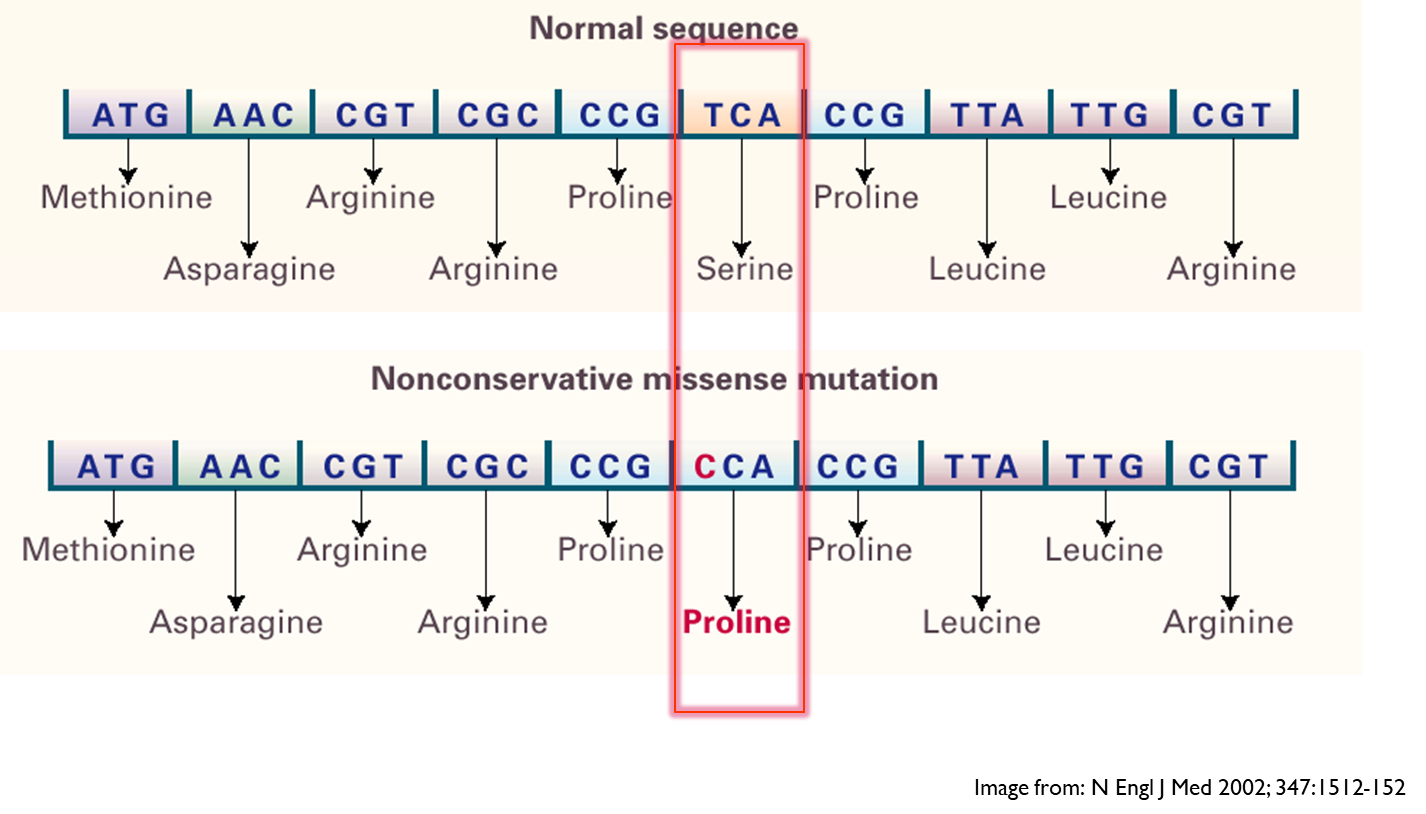
Here the side changes between the two amino acids are very different - likely to have larger effect as it can cause change in shape of protein
e.g. in clinic - beta 2 adrenergic receptor - change in amino acid position 16 caused by SNP which leads to arginine being replaced with glycine resulting in a receptor that has a lower response potential. Doesn’t respond as well to agonists therefore people with asthma with this mutation are less likely to respond to salbutamol
Non Synonymous- Nonsense
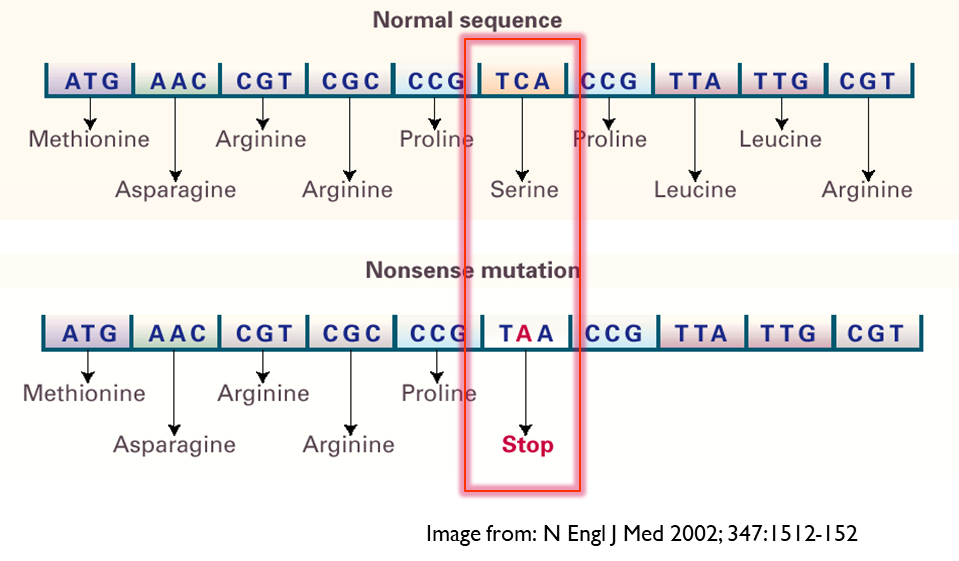
Codon change to STOP codon leading to premature termination of protein - effect on protein depends on where it occurs, will probably have less effect if it happened towards the end rather than at the start
•An example of this is found in an allele of the CYP2C19 gene (*3) - leads to non-functional allele.
•
•
•
•

•The CYP2C19*3 allele is non-functional
How many SNPs have been identified to date?
•Human SNPs are submitted to the NCBI small nucleotide polymorphism database of nucleotide sequence variation (dbSNP)
•Each SNP has a unique identifier (ss)
•The database holds > 720 million unique SNPs (reference SNPs) (Build 154 July 2020)
Base insertion or deletion (InDELs)
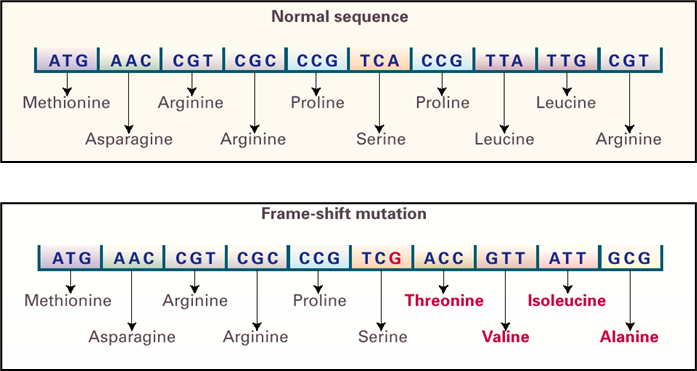
•Used to refer to insertion or deletion Occur due to stand slippage during DNA replication
•+1 frameshift - extra base inserted therefore reading frame moved forward one base - can lead to it being cut short or a much larger polypeptide being formed.
INDELS occur during DNA replication due to strand slippage. If slippage occurs in newly synthesised DNA this leads to an insertion of an additional base which then becomes fixed when the DNA is replicated.
If slippage occurs within the strand being replicated this will lead to deletion of the base within the newly formed DNA.
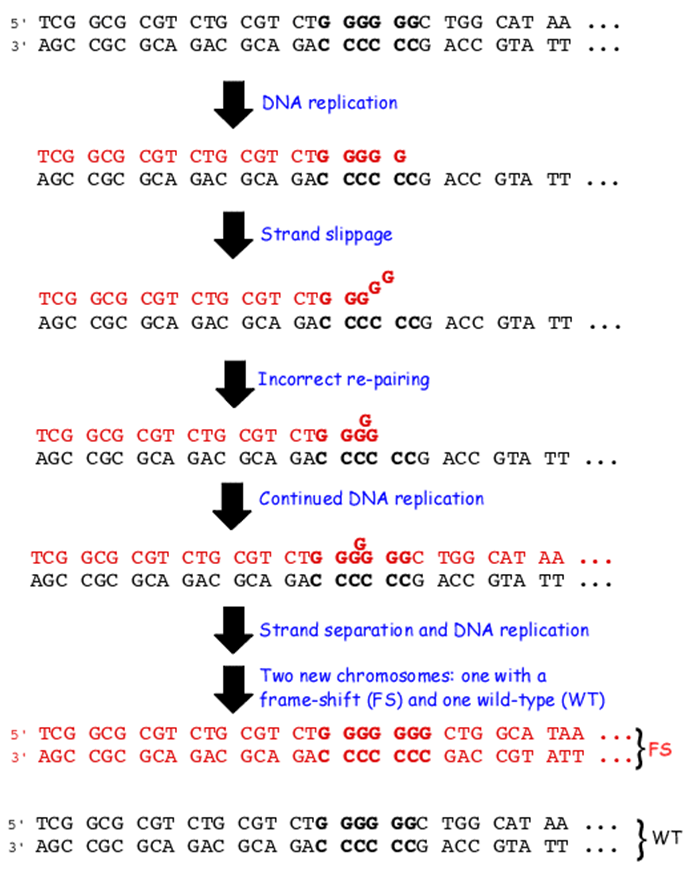
insertion / deletion (Indels)
•90% of indels are1-10 bases long
•Only 1% are of > 100 bases)
Indel examples
Wildtype sequence
ATC TTC CAG CCA TAA AAG ATG AAG TTT
3bp deletion (TAA)
ATC TTC CAG CCA AAG ATG AAG TTT
4 bp insertion (TGTG shown in red)
ATC TTC CAG CCA TAT GTG AAA GAT GAA GTT T
Effect of single nucleotide variations
Normal message
THE BAD AND OLD DOG ATE THE FAT CAT
DNA variations
THE BAD (nonsense, stop codon leads to truncation)
THE BAD END OLD DOG ATE THE FAT CAT (missense)
THE BAD NDO LDD OGA TET HEF ATC AT (deletion A)
THE BAD AAN DOL DDO GAT ETH EFA TCA T (insertion A
Microsatellites
•A type of repeat length variation
•Also known as Simple sequence or short tandem repeats (SSRs / STR)
•Repetition in tandem of a short (2 to 6 base pair) motif with variable copy number across individuals (from 5 to 5,000 times)
•Most are di-, tri-, and tetra-nucleotide repeats repeated 20-50 times
Individual 1 - trinucleotide repeat of 4 repeats
GCGTATACGGGTTACCCCCTTTGCAATCAGTGCAGCAGCAGCAGTGCCAAGCAAAAATAACGCCAAGCAGAACGAAGACGTTCTCGAGAA
Individual 2 - trinucleotide repeat of 21 repeats
GCGTATACGGGTTACCCCCTTTGCAATCAGTGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGCAGGTGCCAAGCAAAAATAACGCCAAGCAGAACGAAGACGTTCTCGAGAA
This type of variation can have an impact on the ADME of chemotherapy drugs
Minisatellite
•Also a repeat length variation
•Also known as variable number tandem repeats (VNTRs)
•14-100 bases long clusters of tandem repeats
•repeating pattern spanning 500 to several 1000 bases, commonly 4 – 40 x per occurrence
•Highly polymorphic across individuals - used for DNA fingerprinting.
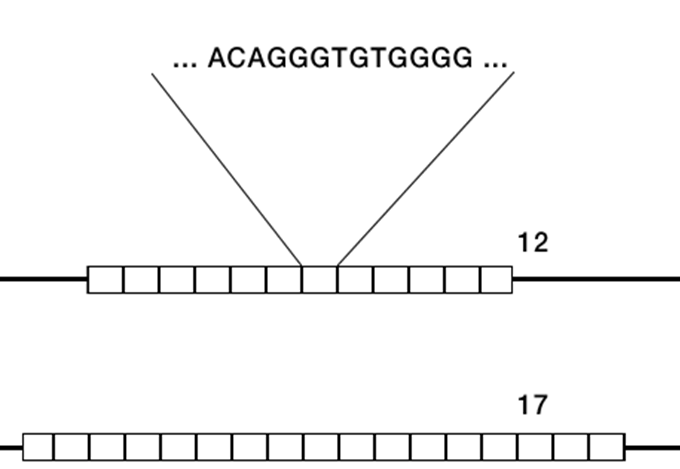
•Role in the regulation of gene expression
•Also used for DNA fingerprinting
Copy number variations (CNV)
•These are larger scale genetic variations and include insertions, deletions, duplications and complex multi-site variants that range from kilobases (1000s) to megabases (100,000s) in size.
•Originally only though to occur in the unstable genomes of cancer cells
•Actually account for ~12% of the genome in stable genetic variations
•CNVs comprise at least three times the total nucleotide content of SNPs.
•Since CNVs often occur in genes, they may have important roles both in human disease and drug response.
A copy number variation that leads to the deletion or duplication of whole gene will influence the quantity of the gene product and can have important roles in both human disease and drug response. CNV explain that ultra rapid metabolization associated with the zip 2d6 isoforms where there are two or more copies of this gene leading to a higher activity of this enzymes. The Star six allele of this enzyme, which is which has a poor metabolised status, is associated with the consequence of a complete deletion of the gene.
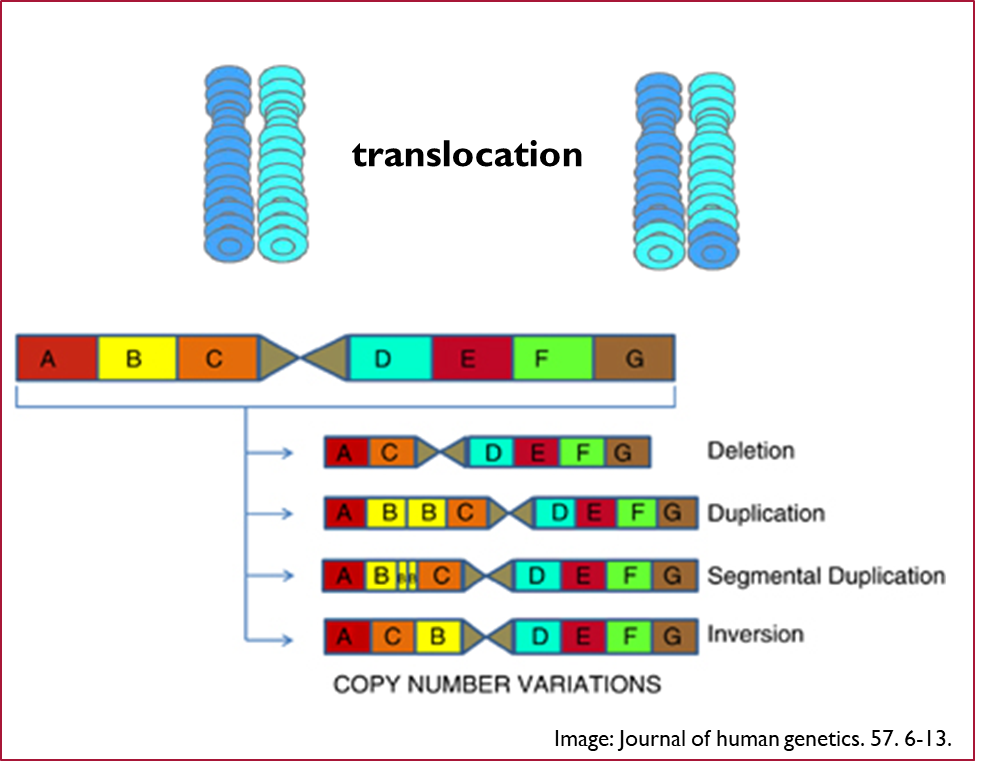
Understanding the consequences of genetic variation
•Inherited variations in the genome combine with environmental factor to produce a phenotype.
•Understanding how genetic variation affects phenotype (e.g. disease predisposition or response to therapy) requires association of the variation with the phenotype response
•This can be achieved by microarray analysis (GWAS) or sequencing of the genome.
•A major issue with this is the size of the genome - need to reduce the number of SNPs to analyse.
•Requires identify a minimum set of Tag SNPs that represent the population under investigation. Its achieved by identifying represented SNPS’s that are the minimum set needed to cover a whole genome, and to use an SNP dataset that is relevant to the population being studied
Haplotypes
•Inheritance of chromosomal DNA is not random, recombination occurs at hotspots.
•Linkage is common in the human population, particularly in genetically isolated sub-populations.
•A group of alleles for neighboring genes on a segment of a chromosome are very often inherited together as single unit is known as a haplotype block
•An SNP in one of these regions can be used as a TAG for the whole region if the inheritance of the “block” as a single piece is maintained
•Linkage disequilibrium is a measure of this inheritance
Linkage Disequalibrium
T and G are SNPs in a gene as are A and C. The two chromosomes are shown
If there is no recombination between these two SNPs. SNP T is inherited with SNP A and acts as a “tag” for it. They are in high LD. The same applies to G and C SNPs.
If there is a recombination site between the two, recombination may or not occur so SNP T does not always tag SNP A. They are in Low LD
Tag SNPs - One SNP may act as a marker for many
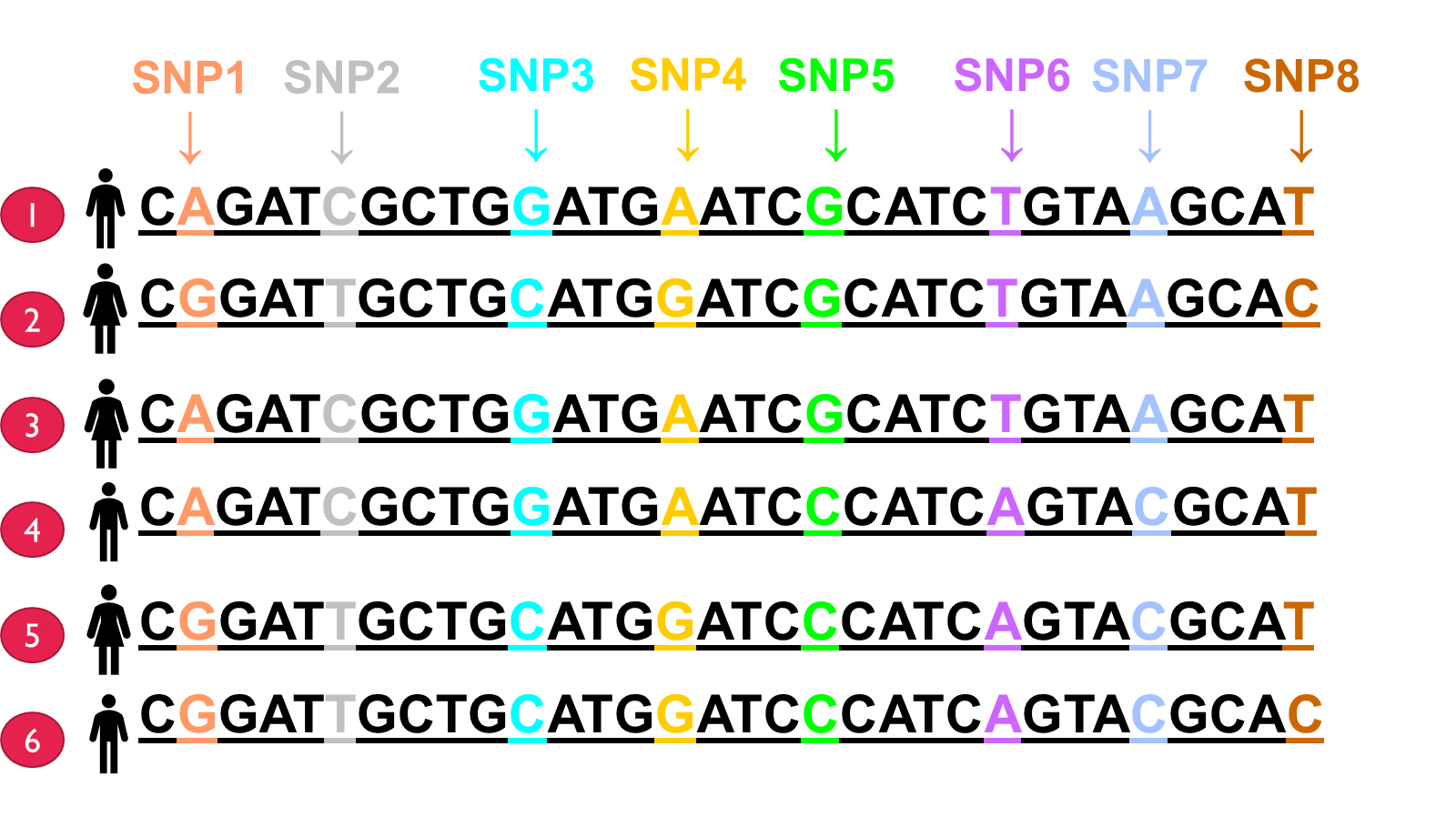
In these 6 people DNA has been sequenced and 8 variations have been found in 10,000 bade region. Here three bases represent the distance between each of those variations. Basically SNP 1 is biallelic as its either A or G, with SNP 2 it will be C if SNP 1 is A and the same until SNP 5 - linkage disequilibrium
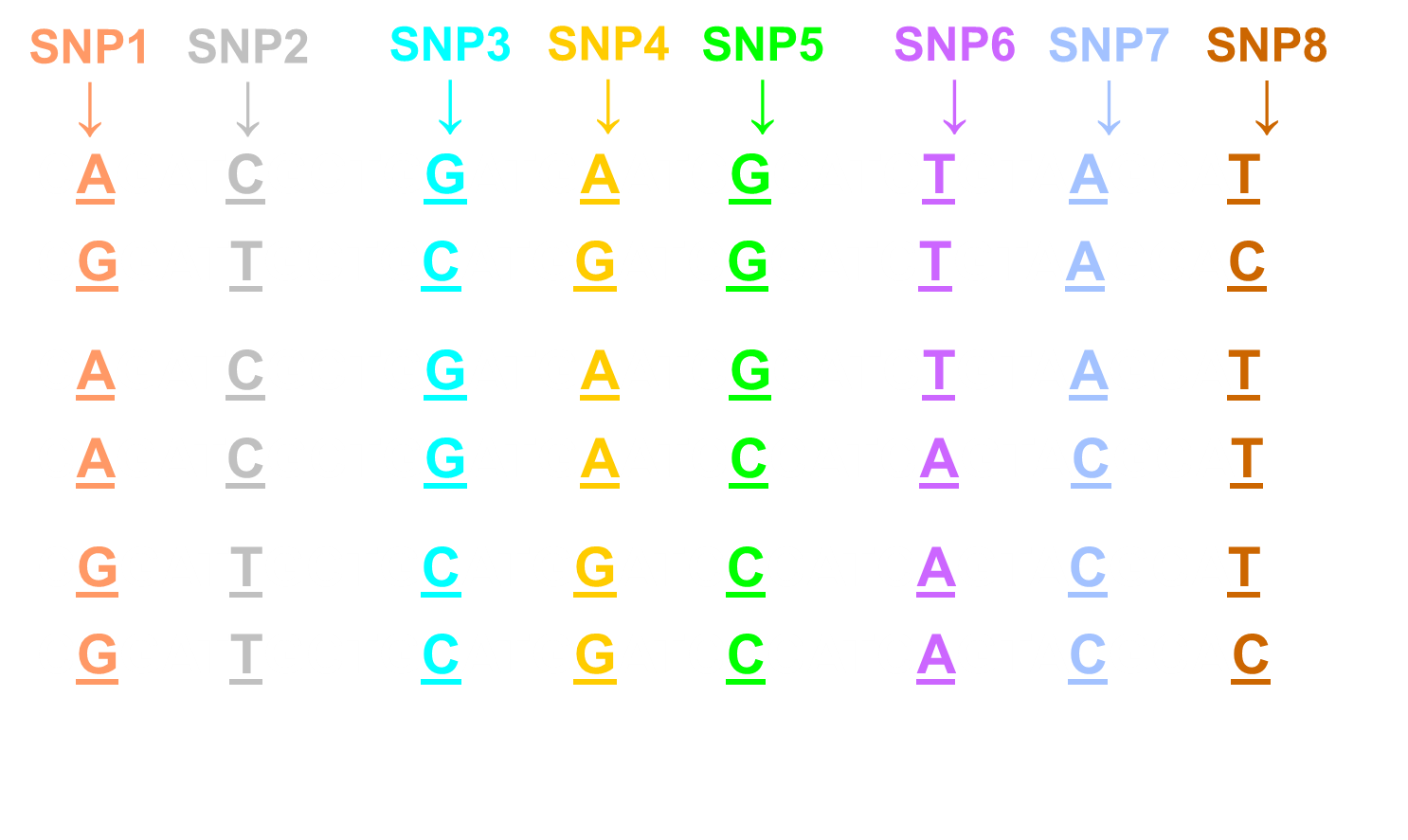
Linkage disequilibrium is then you don’t have random distribution of the variations
cont
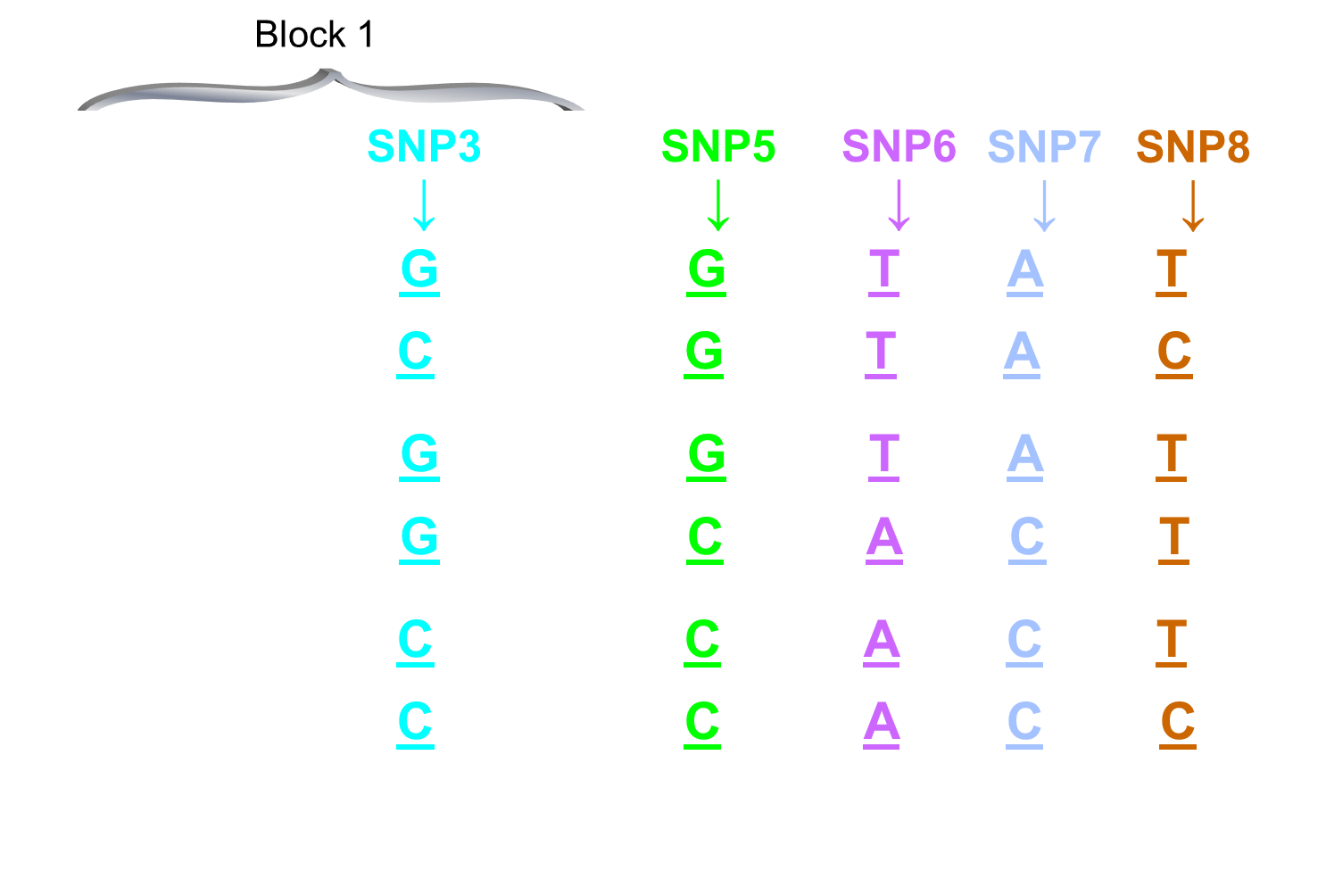
We can use SNP 1 to track SNP 2,3 and 4 but not 5 - first haplotype block
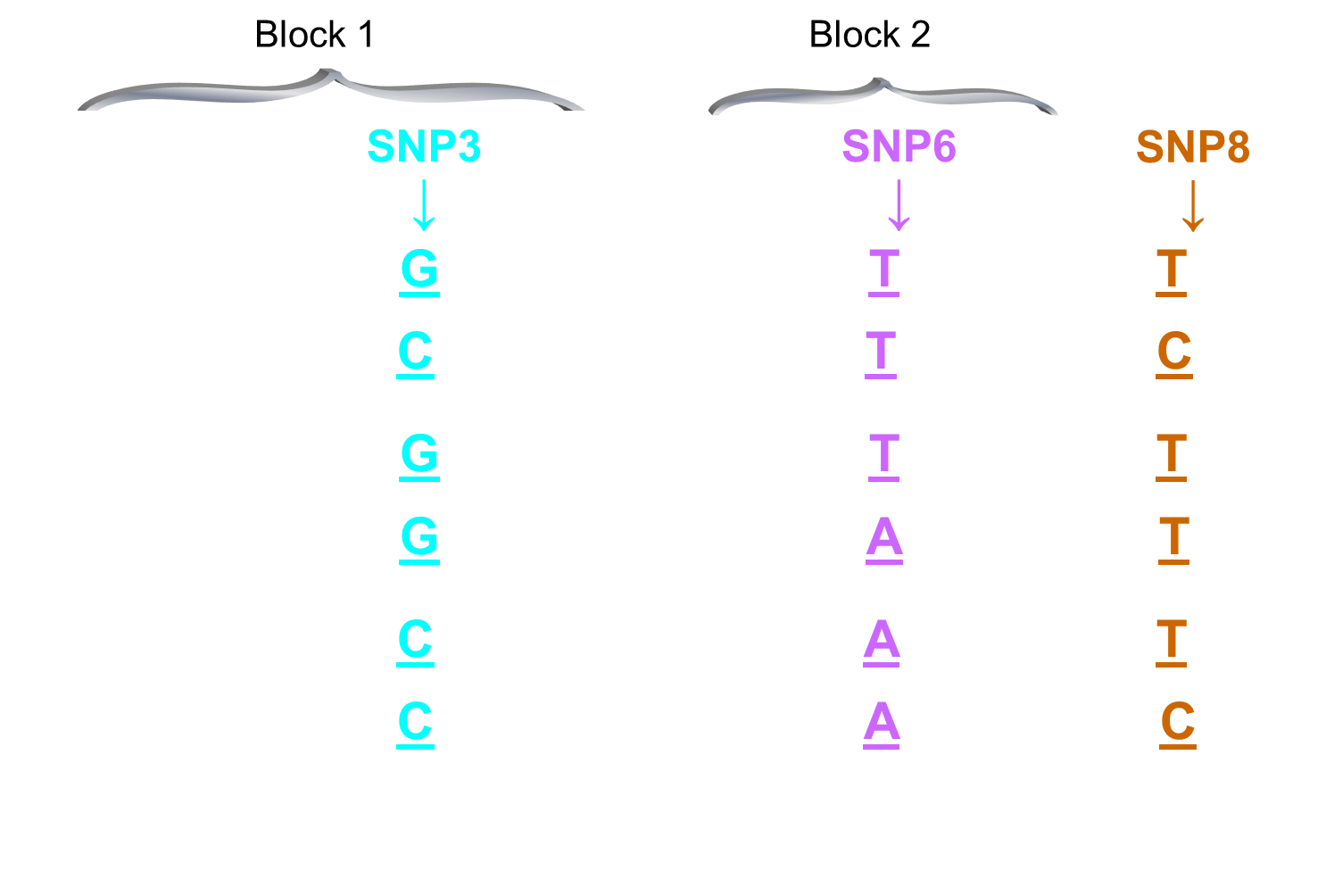
ONE SNP MAY ACT AS A MARKER FOR MANY

Linkage disequilibrium
•There is high LD within haplotype blocks and low LD between blocks or outside blocks
•LD = 100 this is full LD i.e. they are inherited together (very close together),
•LD = 0, no linkage (far apart).
•TAG SNPs are arranged in haplotype blocks that are in relatively high LD - more likely to be inherited than not - think of like BFF’s always together
Mapping the Relationships Amongst SNPs
•This region contains 11 SNPs
•The matrix shows the linkage between each SNP
•This enable a set of TAG SNPs to be identified for use in association studies
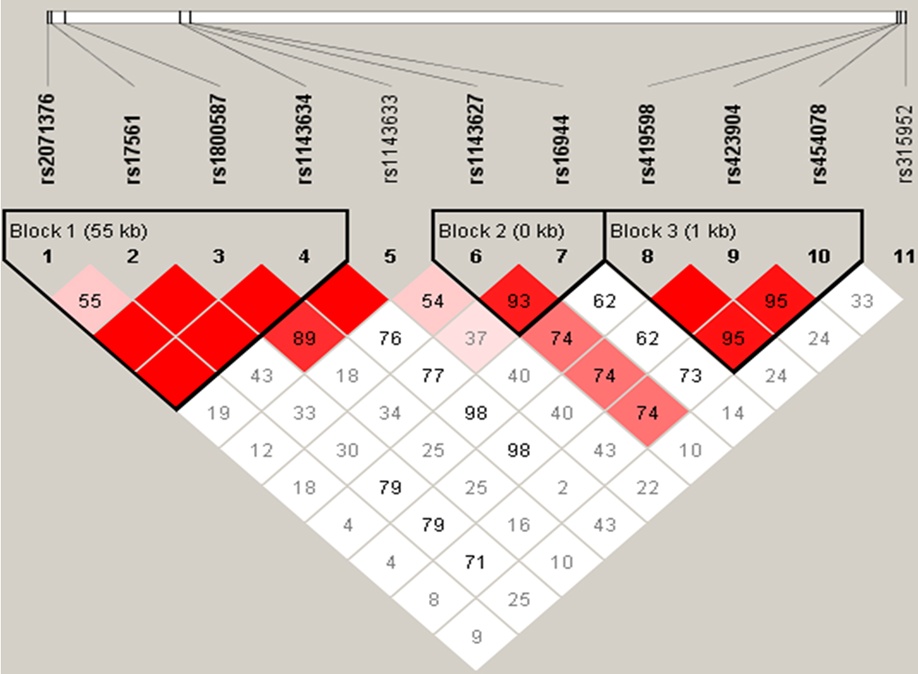
The HapMap project
•The HapMap project was an sequence analysis project aimed at producing a catalogue of genetic variants (single nucleotide and copy number polymorphisms) within the human genome from diverse global populations
•Project outcomes:
•investigated variations within various sub-populations
•collated data from other sources
•tested linkage between variations in populations.
Practical use of Hapmap project data
•Defines the set of SNPs that will cover an ethnic group’s genome (and common SNPS across ethnic groups)
•It has enabled population specific analysis (same ancestry) to:-
•Identify genetic variations prevalent amongst diverse races that define drug responsiveness
•Identify how population diversity has an impact on disease susceptibility
•Enable the generation of population specific genetic test
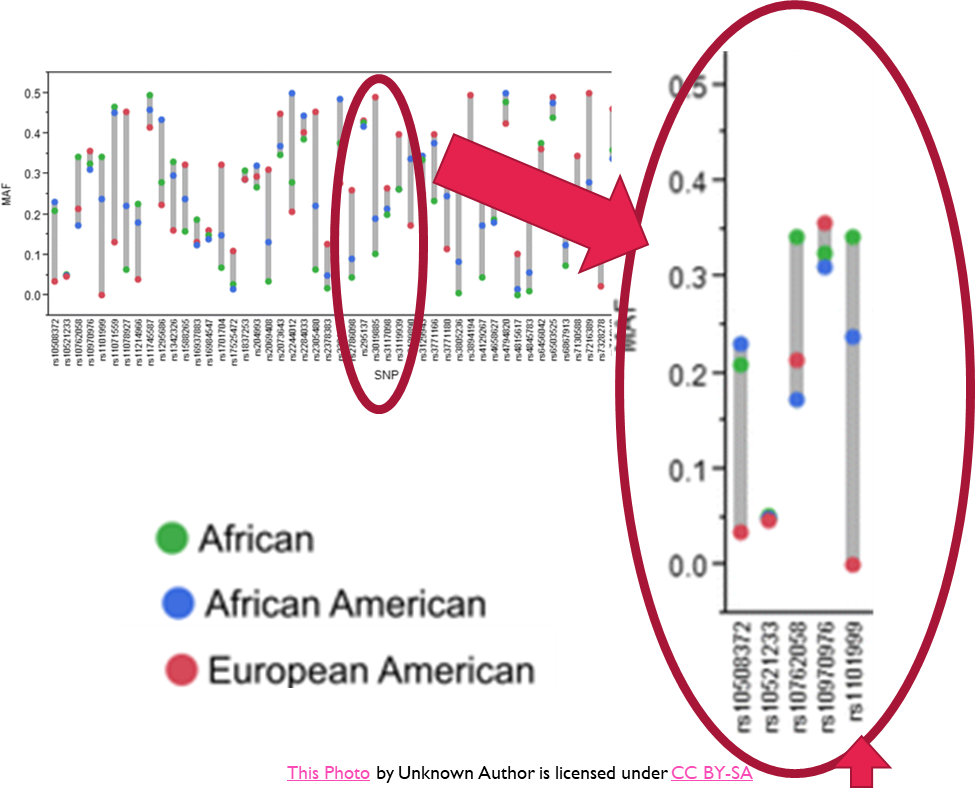
findING the link between genotype and phenotype –
Using TAG SNPs In A GWAS
•Using TAG SNPs reduces the number that need to be screened across the genome to ~500,000 for a population group

•Those with the phenotype all have an ‘A’ at position 4 in Seq X, while those without have a ‘T’
•Seq X does not have to be a gene associated with the response but needs to be inherited with it.
•Seq X is a biomarker for the phenotype