Hemoglobinopathies
1/39
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
40 Terms
Why are hemoglobinopathies important?
Most common group of genetic disorders in the world
Affect many millions of people (carrier frequencies of up to 20%)
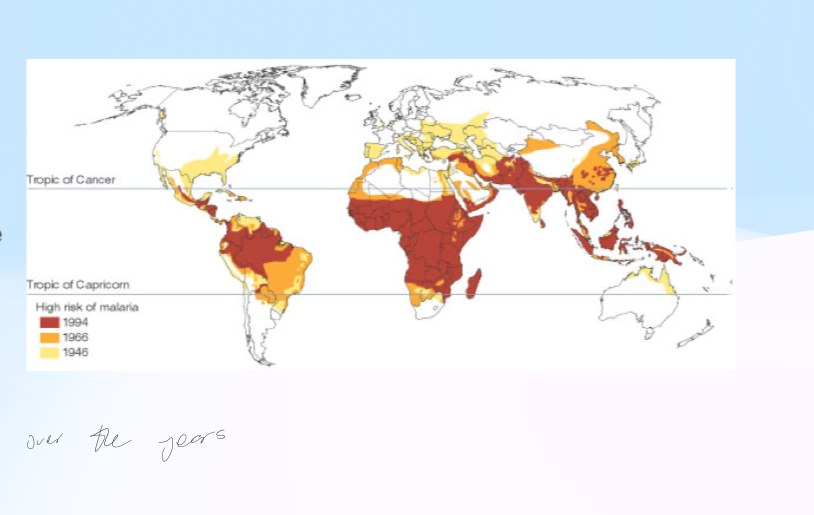
Affects people all over the world, although concentrated in the “malaria belt”
Chronic problems that take a huge toll on individuals, health care systems, economies etc
What do the 3 hemoglobinopathies have in common?
Effects = Hemolytic anemias (RBCs lyse). Circulation problems (sickle cell)
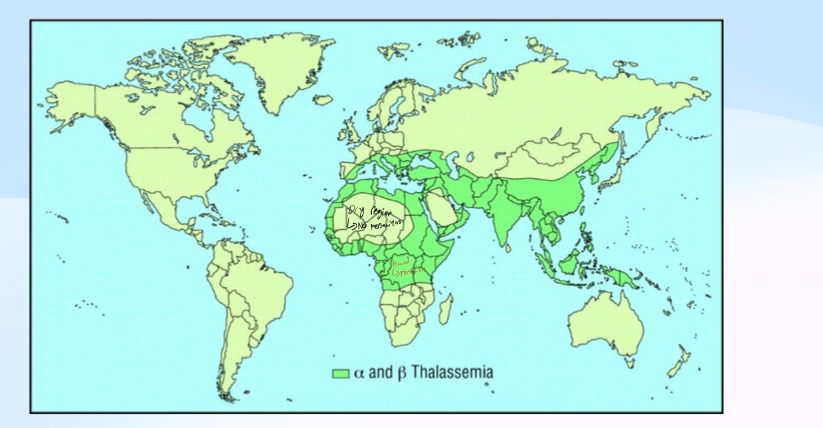
Distribution = Matches current (or historical) distribution of malaria. High in West Africa, Middle East, Indian subcontinent, China etc. Historically low in higher latitudes (where theres no malaria)
Selection = High carrier frequencies due to heterozygote advantage with malaria (Plasmodium) infection. Hemolytic anemias interfere with parasite that hides inside RBCs
Whats the distribution of Thalassemias?
Whats the distribution of Malaria?
What causes hemoglobinopathies?
Disorder caused by mutations in the globin genes that give rise to hemoglobin
Hemoglobin is the protein that transports oxygen from the lungs to our tissues
What’re the 3 hemoglobinopathies?
Sickle cell disease
Beta-thalassemia
Alpha-thalassemia
What is Hemoglobin (Hb)
Hetero-tetramer = 4 subunits, two each of different globin proteins plus an iron containing heme ring. Many Hb tetramers contained within each red blood cell
Whats the structure of hemoglobin important for?
It’s important for its stability, solubility, and oxygen affinity (should be high enough to collect in blood, low enough to release in tissues)
What can abnormal Hb structures cause?
Abnormal Hb structure can cause insoluble or unstable Hb and lead to hemolysis (breaking of the RBC). This lowers RBC count in the blood
Abnormal structure also changes oxygen affinity, so Hb doesnt effectively carry oxygen to tissues
What does reduced Hb result in?
Reduced Hb results in small RBCs and reduced oxygen capacity
Which globin genes are featured in both fetal and adult?
Alpha1
Alpha2
Which globin genes are featured only in the fetus?
Gamma genes
Which globin genes are only featured in adult?
Delta
Beta
Explain what makes up fetal hemoglobin (HbF) and what it is
Made of 2 alpha and 2 gamma chains which are expressed in fetus
In the first year after birth gamma is replaced with beta globin to give HbA (2 alpha and 2 beta chains)
Some HbF may persist in adulthood due to persistent gamma globin expression but this varies between individuals
Explain all the adult hemoglobin’s (HbA, HbA2, HbF)
HbA = 2 alpha and 2 beta
HbA2 = 2 alpha and 2 delta
HbA represents about 97% of adult Hb
Some HbF may persist after birth
Presence of other Hbs indicates a problem with the balance of expression of the globin genes
Whats sickle cell disease?
Autosomal recessive blood disorder
Abnormally-shaped RBCs that can block blood vessels and lead to loss of circulation, organ damage
Hemolytic anemia cuz it leads to break of RBCs but since sickle cell can also blood vessels, which isn’t in thalessemia
Whats the mutation of sickle cell disease?
All people with sickle cell trait or disease have the same disease-causing missense allele
NOT a cause of allelic heterogeneity
Haplotype analysis of current humans suggests this allele has arisen only 3 times (low mutation rate cuz its a specific bp substitution)
Any regular lof mutation in the beta globin gene would cause beta thalassemia
Whats the mechanism of sickle cell disease?
Mutation: missense mutation that results in a hydrophobic amino acid on the surface of a globular protein. Glu-Val substitution (acidic, hydrophilic to non-polar, hydrophobic.) This can cause proteins to aggregate (hydrophobic residues will prefer to stick to each other rather than be exposed to water)
Hb is already a tetramer. So the sickle cell mutation results in elongated aggregates of Hb (called HbS). This causes the sickle shape of the RBCs
What’re the SCD effects on circulation?
Sickled RBCs are more likely to block blood vessels. More likely during hypoxia or dehydration
Explain the heterozygotes for sickle cell trait
People who are heterozygous for the sickle cell allele have a mix of normal and abnormal beta globin and a mix of normal (HbA) and abnormal (HbS) hemoglobin proteins in their RBCs
RBCs tend to be normal shape unless oxygen levels drop
Hypoxia can cause some suckling of the RBCs and episodic circulatory problems so it can be a problem for athletes or people who have respiratory problems, go to high altitude
Explain sickle cell distribution
West Africa, Mediterranean, Middle East, India
Explain heterozygote advantage in sickle cell disease
+/+ = Normally perfectly healthy but not when malaria, when malaria they are selected against
± = Best cuz don’t have issue with sickle cells and have immunity against malaria. In time, this will become the most common allele from cuz they have no selection against
-/- = Selected against cuz sickle cell
Explain balanced selection in heterozygote advantage
Heterozygote advantage can cause the increase in frequency of a disease allele in a population Even if homozygotes are at a disadvantage and this frequency will remain high as long as the non-genetic selection (ex: malaria) is present
If the non-genetic selection is no longer there. Frequency will decrease, but only very slowly and over many generations. This is because most disease alleles are held by the carriers and there’s no disadvantage for them, this applies to SCD and the thalassemias
What’re some treatments for SCD?

Whats Beta thalassemia?
Happens cuz only one beta globin gene in Ch 11 cluster
Caused by lof mutations in beta globin (theres allelic heterogeneity). These are often deletions. There can be complete or partial loss of function alleles (allelic heterogeneity)
Reduction in beta globin causes imbalance between alpha and beta globin and reduces the amount of HbA produced
What’re the 3 different types of beta thalassemias?
Beta thalassemia minor = heterozygotes
Beta thalassemia intermedia = homozygous but not complete lof
Beta thalassemia major = homozygous complete lof
What inheritance does beta thalassemia have?
Autosomal recessive inheritance
Explain the heterozygotes of beta thalassemia
Heterozygotes have thalassemia minor like sickle cell
RBCs may be smaller than normal due to less hemoglobin
Reduced amount of beta globin results in reduced HbA levels
Not usually a clinical problem. But they’re carriers and have a risk of affected children
Explain homozygotes of beta thalassemia
Beta thalassemia intermedia and major
Anemia, skin pallor, bone deformities, cardiac arrhythmias, hepatomegaly, jaundice
Beta thalassemia intermedia has the same symptoms but less severe. Treatment depends on severity. Might include increasing Hb or RBC production, folic acid, hydroxyurea etc. Might include transfusions as needed
What’re the treatments for beta thalassemia?
Regular blood transfusions are required for survival
Also usually need iron chelators to prevent iron overload from the transfusions cuz every blood transfusion adds iron to the system. An iron chelator binds to iron in the blood and facilitates removal via the kidneys
Patients with TDT and >age of 12 may be eligible for gene therapy
Beta thalassemia major is also called transfusion dependent thalassemia (TDT)
Whats alpha thalassemia?
Many sub types
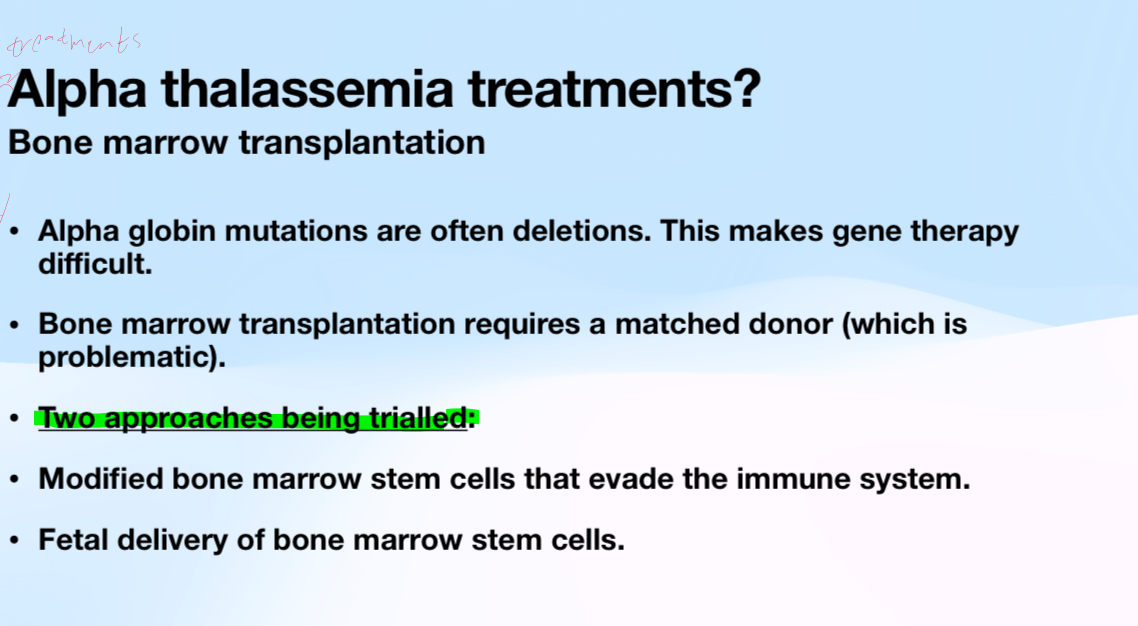
There are 2 alpha hemoglobin genes (4 alleles in genome in total). A person with alpha thalassemia can have 1,2, 3,4 lof alleles. There’s are often deletions of one or both genes (allelic heterogeneity)
What’re the different types of alpha thalassemia?
1 lof alleles = alpha thalassemia silent. No clinical phenotype
2 lof alleles = alpha thalassemia trait. Mild anemia
3 lof alleles = alpha thalassemia intermediate. Hemoglobin H disease
4 lof alleles = alpha thalassemia major. Generally do not survive to birth. Condition called hydros fetalis (fetal edema). Can be treated with intrauterine transfusions. Hb Barts in high proportion
Explain alpha thalassemia intermedia (HbH disease)
Typically 3 or 4 alleles are defective (usually deleted)
Tetrameric beta globin (HbH) forms due to excess beta globin. Low solubility of HbH leads to inclusion bodies and splenic destruction of the RBCs
Can also be developmental issue due to reduced HbF levels
Treatment depends on severity. Folic acid to assist with RBC production, transfusion if theres an episodic event. Possible bone marrow or stem cell transplantation
Explain the Hb barts diagnostic for alpha thalassemia
Tetrameric gamma hemoglobin (reduced alpha globin)
Relatively insoluble Hb = easy to identify in electrophoresis
High affinity for oxygen so doesnt release it to tissues
In newborns, Hb barts indicates mutation in at least one alpha globin genes
What’re treatments of alpha thalassemia?
Same as other hemolytic anemias
Iron supplementation isn’t indicated
Boost RBC production
Severe symptoms could lead to transfusions, splenectomy (to best RBC counts, bone marrow/ stem cell transplantation)
No gene therapy available at this time for alpha thalassemia
Explain alpha and beta co-inheritance
Because both can be common in the same region, patients may carry mutations in both alpha and beta globin
Provided they aren’t complete lof conditions, mutations in both can partially compensate for each other by restoring balance between alpha and beta chains
Ex: beta thalassemia intermedia may be less severe when there is a reduction in alpha globin production
Whats the prevention of alpha thalassemia?
Regions with high carrier frequencies typically do carrier screening to reduce likelihood of carriers for the same condition mating
Prenatal or newborn screening to identify individuals with SCD or major or intermideia forms of thalassemias. Early intervention has better outcomes. Screening can be done with electrophoresis of Hb (from a blood sample)
What’re some new gene therapy treatments for hemoglobinopathies?

Explain the potential for bone marrow transplants for alpha thalassemia and the 2 approaches
Understand the clinical case