pmb 114 mt3
1/99
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
100 Terms
Rabies (genome + disease)
(-)ssRNA enveloped virus, rhabdoviridae
Severe acute progressive encephalitis
transmitted via saliva through bite
neurotropic (attacks neurons), NOT in blood
virus travels to the brain via retrograde axonal transport
nearly 100% fatal in humans
Rabies (prevention)
Vaccinate the dogs!
bats also have it
Prevention = vaccination
- pre exposure prophylaxis (vaccine) given to high risk people
- Post exposure prophylaxis (PEP) (vaccine) given post bite before symptoms
No treatment once symptoms develop...
Cryptic rabies cases
associated with silver haired & easter pipistrelle bats
they have super small bites --> unnoticed
enhanced virulence + enhanced infectivity bc superficial route
they replicate in non neural tissue and at lower temps
(Sin nombre) Hantavirus
Hantaviridae
(-)ssRNA enveloped virus
causes hemorrhagic fever with renal syndrome (HRFS - old world)
hantavirus pulmonary syndrome (HPS - new world)
rodent host, people usually get it in camping locations
shed through urine/feces, gets aerosolized & inhaled
Infects epithelial cells, lungs can fill up with fluid --> Endothelial cells become "leaky"
in shrews and bats
Retroviruses (genome + virion structure)
(+)ssRNA
(+)ssRNA --> DNA intermediate (RT) --> mRNA
polyprotein method
env, gag, pol
env = envelope - surface + transmemnbrane
gag = core - protease, matrix, nucleocapsid, capsid
pol = enzymes - integrase, RT
Translation is NOT first, so protein has to be deposited into cell with genome
Not a segmented virus, "diploid" = 2 identical copies of viral genome
Retroviruses (simple vs complex)
simple = rna tumor simple genomes: Encode only unspliced and singly spliced mRNAs,
complex retroviruses encode multiply spliced mRNAs
Retrovirus Replication
Binding receptors on cell surface, fuse w plasma membrane to enter
RT of viral RNA in the core by RT --> dsDNA
core/capsid is starting to dissolve otw to nucleus
dsDNA and integrase enter genome (pre-initiation complex)
integrates enzyme into cellular genome → becomes provirus
Forever integrated into genome
Virus makes new progeny viruses through RNA poly 2 (host) or translation of spliced mRNA
proteins translated as polyproteins --> immature virus particles assembled and released via budding
maturation after virus released
Protease mediated Gag and GagPol processing causes major change in virion morphology
Gag made from gag-pol due to ribosome frameshifitng
Retrovirus Reverse Transcriptase (2 enzymatic activities
Polymerase Activity:
- Primer dependent (needs DNA or RNA)
- Template can be DNA or RNA
- Only incorporates dNTPs not rNTPs (only makes DNA but can use either as a template)
RNAseH acvitity:
- cleaves RNA when in duplex form (RNA:RNA, RNA:DNA)
error prone
Reverse transription process
RNa --> dsDNA
RNA has R for repeat, unique 5' end sequence (U5) , primer binding site (PBS)
3' end = polypurine tract, 3' sequence unique (U3), another copy of repeats
RT turns ssRNA into dsDNA --> ends are different from template, they have long terminal repeats which contain U3, R, and U5 (in that order)
dsDNA integrated, U3 has promoter --> transcription starts at R, rna transcript corresponds to genomic copy
retroviruses (rna to dsDNA details)
go watch the video LOL
cellular tRNA bound to PBS of rna = orimer to initiate reverse transcription (RDDP)
viral RT RNAseH degrades dna hybridized to DNA
newly synthesized DNA transferred and hybridized to repeat region at end of genome (where the tetris S shape comes from)
full length genome is copied by rev transc (RDDP)
viral RT RNAseH degrades RNA hybridized to DNA, except PT region (resistant)
DNA synthesis resumes using hybridized ppt RNA as primer (makes 2nd strand) (DDDP)
viral RNAseH degrades ppt primer and releases tRNA (now hybridized to DNA)
PBS primer hybridizes to complementary sequence at the start of newly synthesized DNA
both - and + DNA strands extended to create full lendth dsDNA genome for integration
Retrovirus Integration
integrase is in core, enters nucleus and is entered into target DNA sequence
duplication of host target sequence lets you see where it is
RNA poly transcription is only way out of the genome
HIV/AIDS (genome + transmission + immunity + origin)
(+)ssRNA
transmission as cell associated virus or free virus via bodily fluids: blood, sex fluids, breastmilk
13% infected don't know they have it
Most diagnoses made in early 20s, or 13-24
CCR5∆32 mutation are resistant to HIV infection --> can donate blood to help infected individuals
SIV, primate to human origin
HIV/AIDS pathogenesis (cellular)
Targets CD4⁺ T cells due to specific receptor interactions
Env gene encodes a glycoprotein → cleaved by viral protease into:
gp120 (SU): binds CD4 receptor
gp41 (TM): facilitates membrane fusion
gp120 binds CD4 → triggers conformational change → exposes coreceptor binding site
Coreceptors = CCR5 or CXCR4 (co-receptor is required for entry)
Virus may prefer one (CCR5-tropic or CXCR4-tropic)
After coreceptor binding → exposes gp41 transmembrane domain
gp41 is a class I fusion protein: promotes fusion of viral envelope with host cell membrane
Result: Viral RNA enters host T cell → infection begins
HIV/AIDS (t-cell infection)
Cytotoxic T cells (CD8⁺)
Interact with MHC I on infected cells
Kill infected cells directly
T-helper cells (CD4⁺)
Interact with MHC II on antigen-presenting cells (APCs) like dendritic cells, macrophages, B cells
Activate other immune cells, including:
B cells → become plasma cells → make antibodies
Cytotoxic T cells → enhance killing
HIV selectively infects CD4⁺ T-helper cells
Impairs both adaptive and innate immune response
MHC II signaling is disrupted → B cells can't activate → poor antibody production
Outcome:
Immune system can't respond to new threats
Leads to AIDS: death is usually due to secondary infections or cancers
HIV/AIDS infection
AIDS = your CD4 T-cells drops below a certain amount
Initially: HIV replicates immediately (integrating into the genome occurs, permanent)
Viral load stabilizes, virus still replicates actively but host immune system keeping it under control (good)
bad --> HIV wins, t cells decline until no immune response can be sustained, HIV replicates out of control, host subject to tons of infections.
CD8 T-cells get "exhausted" and die
10^10 virions per day made and destroyed
Increased likelihood of mutation in a virus that lets it escape immune system
rapid vs non rapid regressors (HIV/AIDS)
Rapid regressors are people who develop AIDS very fast
Non-progressors: maintain normal CD4 count at low levels for 10yrs without antiretroviral therapy (ART) (rare) not associated with attenuated virus
HIV/AIDS treatment
ART = antiretroviral therapy, works to keep virus levels low, not knock it out
PrEP and PEP:
PrEP = pre exposure, given to high risk individuals
Consistent use of it can reduce risk by 99%
Key for preventing virus from integrating
PEP = post exposure
Should be started within 72 hours
^ not effective if symptoms already occur
Access to these is super unequal
HIV/AIDS inhibitors
Attachment inhibitors:
Binds to GP120 surface of spike protein to prevent interaction with CD4
Mutations overcome it tho
Post attachment inhibitors:
Antibodies that bind once receptor binding domain binds with CD$, stops CD4 conformational changes
CCR5 antagonists
Molecules that bind to it and block interaction with GP120 after CD4 interacts
This wouldn't work for mutated individuals
Fusion inhibitors:
Fusions allows gp41 to insert into membrane and fold back on itself for fusion
Folding back requires interaction between sub domains HR1 and HR2
Took small fragment of hr2 and made a peptide of that which mimics HR2 so HR1 binds to it, then HR1 cant fold back on itself
separate? Lenacapavir drug:
for prep, Lenacapavir binds to hexamers so progression through breaking capsid can't happen
HepB (RT activity)
dsDNA, uses RT
Viruses enter cell with ds DNA → becomes + RNA → proteins or - DNA to become dsDNA
RNA template to dsDNA is where RT works
You get a partially double stranded genome
Enzymatic activities of RT:
RNase activity (Cleaves)
Polymerase activity (RNA/DNA temp → makes DNA only)
Hep B P Protein
P protein in HepB has three key domains:
Terminal Protein – primes DNA synthesis by acting as a protein primer.
Reverse Transcriptase (RT) – copies viral RNA into DNA.
RNase H – degrades the RNA strand of RNA-DNA hybrids during reverse transcription.
referred to a lot as just hepB’s RT
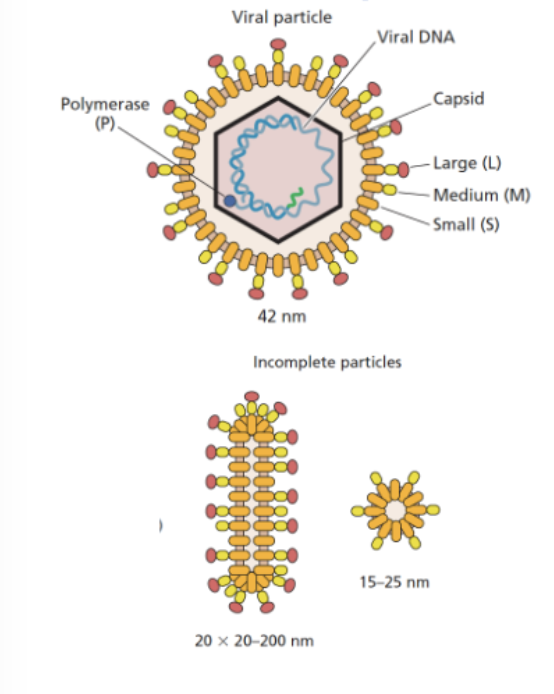
HBV particles
Dane particles: They contain the viral envelope, capsid, and the HBV genome (partially double-stranded DNA).
HBV makes huge numbers of non-infectious particles (envelope only)
Decoy particles distract immune system
Helps real virus evade detection and persist in the host
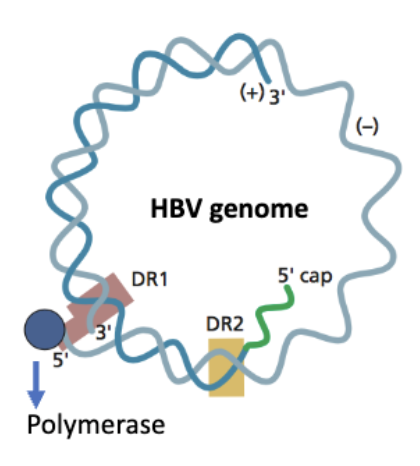
hep B genome
atypical: partially double stranded
Polymerase (P protein) is covalently attached to the 5′ end of the (–) strand
(+) strand has a 5′ cap, like eukaryotic mRNA
🔁 Both strands contain Direct Repeats (DR1, DR2)
These are short, identical sequences at specific sites
The 5′ ends of both DNA strands are near or within these direct repeats
DRs are important for genome replication and circularization
RT does RNA to dsDNA, happens in confines of capsid → RT activity runs out of dNTP’s can't finish job → partially dsDNA molecule with some leftover RNA
RT in hepB vs retrovirus (dNTPs)
Retrovirus: RT activity happens in capsid as its moving to the nucleus, capsid disassembly and brings in dNTPs
Hep: reverse, RT as capsid assembled, runs out of dNTPs
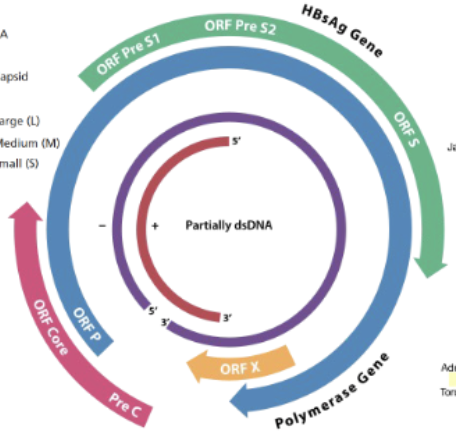
HBV overlapping genes + consequences
4 of them (overlapping ORFs)
PreS1/s2 ORFs = surface antigen, makes S,M,L antigen proteins
ORFcore PreC = makes capsid
ORFP = encodes viral polymerase (P)
orf X = transcriptional activator (irrelevant?)
Overlapping genes = if theres a mutation in 1, it can mess up multiple (mutation in S can ruin P for ex)
or can mess up the polymerase
HBV replication cycle
Virions use receptor exclusively found in liver cells (liver tropic)
Receptor mediated endocytosis
HBV: Formation of cccDNA
Viral capsid delivers partially dsDNA to nucleus
Host enzymes repair DNA → cccDNA
cccDNA = stable, circular, viral DNA reservoir
Does NOT integrate into host genome (unlike retroviruses)
Acts as template for viral RNA + genome production
Host rna poly comes and transcribes mRNA and pregenomic RNA
(pgRNA = template of rna converted to dna that gets packaged into virion)
HBV recycling pathway
Feature | Purpose |
|---|---|
pgRNA + capsid in cytoplasm | Reverse transcribed to dsDNA |
Capsids return to nucleus | Make more cccDNA early in infection |
Envelope proteins missing | So virions aren’t secreted yet |
10–30 cccDNA per cell | Boosts viral transcription + antigen expression |
Liver cell division | Recycling maintains infection (no integration) |
HBV gene expression / leaky scanning
HBV Gene Expression + Leaky Scanning
RNA Pol II transcribes pgRNA + mRNAs from cccDNA
Transcripts have repeats → help reverse transcription
Leaky scanning lets ribosomes skip weak start codons
→ Produces multiple proteins from one mRNASome are truncated or alternative-frame proteins
Efficient way to control protein ratios (stoichiometry)
How HBV makes its relaxed circular DNA genome
DR - direct repeats
P binds ε on pgRNA → primes short DNA
Template switch: P moves to 3′ end
TP primes (–) DNA synthesis (RDDP)
RNAse H degrades RNA, except capped DR2
DR2 RNA primes (+) strand DNA (DDDP)
(+) strand goes DR2 → DR1
DR1 hybridizes to 5′ DR1 to circularize
(+) strand elongates → relaxed-circular DNA
⚠ Incomplete synthesis likely due to dNTP shortage
HBV spread + vaccine
mother to child
intravenous blood transfusions (before screening)
sexual transmission
IV drug use
needle sticks in hospitals
Can be acute or chronic infection
Acute: jaundice, high viral protein levels, immune kicks in and recovers
Chronic: proteins continually produced, immune system fails to clear viral DNA
Likelihood of chronic vs acute is due to age
High risk for children, vaccine happens at birth
Individuals with chronic also have cirrhosis or liver cancer
hepB doesnt cause cell death, it messes up ur liver superbad
if infected, not curable
vaccine given at birth
Arenaviridae (genome + general)
enveloped, bi-segmented RNA genome:
L segment: larger
has Z protein: lays a central role in the virus's assembly, budding, and replication regulation.
S segment: smaller
has IGR - intergenic region (loop) that separates ± regions
nucleocapsid coats RNA
old and new world
cause hemorrhagic fevers
some are non pathogenic
transmitted through mastomys rats —> rat to human —> human to human
urine/feces, or bodily fluid/blood transmission
can be in mammals and snakes
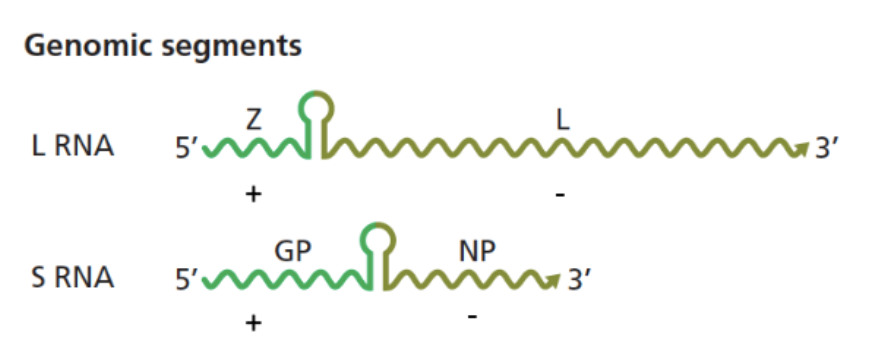
Arenavirus genome expression
Replicates in the cytoplasm.
Ambisense RNA genome: Each segment (S and L) has two genes in opposite directions.
Needs to make:
mRNAs for translation (to make proteins),
Antigenomes as intermediates for replication.
Transcription Phase (Early Infection):
L protein (viral RdRp) binds the 3′ negative-sense ends of genome segments.
It transcribes subgenomic mRNAs for:
NP from S segment
L protein from L segment
These transcripts stop at a central intergenic region (IGR) — a structured RNA stem-loop that signals transcription termination.
Replication Phase (Later):
Once there's enough NP and L protein made:
Full-length antigenomes (complementary to the genome segments) are synthesized.
Then, the 3′ ends of the antigenomes are transcribed into subgenomic mRNAs for:
GPC (glycoprotein precursor → GP1 & GP2) from S segment
Z protein from L segment
timing of protein production (early/late) - arenaviridae
early: polymerase, nucleocapsid proteins
late (after viral rep): Z protein, glycoprotein
Arenaviridae replication & 3’ utr region & no polyad
Arenaviruses use ambisense RNA segments to control timing of gene expression — early proteins (NP, L) come first, then late structural ones (GPC, Z) after replication begins.
The 3′ UTR formed from the IGR helps control when and how much protein is made by influencing translation of viral mRNA — a key regulatory strategy for timing viral protein expression.
Arenavirus mRNAs are capped but not polyadenylated.
To make up for this, the virus relies on regulatory sequences in the 5′ and 3′ UTRs of each mRNA.
These sequences influence mRNA stability and translation, explaining why more NP (from strong UTRs) is made than GP (from weaker UTRs).
Reporter gene experiments confirm this difference in expression is driven by the UTRs themselves.
Arenavirus cap snatching + Z in viral assembly
cytoplasm, cap snatching occurs here
host derived sequences at 5’ end (characteristic of cap snatching)
carried out by 1 protein
late in infection when nZ produced —> time for packaging
Z plays a role in taking virus from translation to assembly, and also
in viral assembly: interacts with glycoprotein GP, nucleoprotein NP, Polymerase L
Z also dampens innate immune response
Z protein from pathogenic can evade immune, non pathogenic cant
Upregulates IFNs to get to antiviral state
Nucleocapsid protein and immune evasion - arenavirus
NP supresses type 1 IFN productoin (z does this too)
Crystal structure of LASV NP shows unique RNAse domain specific for dsRNA —> if it cleaves it , it cant be detected
Reovirused (Genome + general)
REO = respiratory enteric orphan virus
non-enveloped, icosahedral capsids
liner dsRNA genome
double layered capsid
replication in cytoplasm
incomplete uncoating of virions, which possess all the
enzymes required for ds RNA transcription
fecal/oral transmission
Reovirus entry and uncoating
Multiple steps bc of multiple layers
Transmitted fecal-oral, replicate in gut (acidic)
Uncoating happens bc low pH environment of gut
Binds with sialic acid or JAM-A (don't need to know names) receptor mediated endocytosis
Viral particle is double layered → acid induced proteolytic processing of outer capsid → creates intermediate subvirion particle (ISVP), has enzymatic activity, active when outer capsid protein taken away
Structural changes to capsid during entry - Reovirus
Virion → ISVP → core
Think of reovirus like a locked canister:
First, the shell is dissolved in the cell (sigma 3 removed),
Then the docking arm pops out (spike extends),
After docking, the lid of the canister twists open like petals (lambda 2 opens),
Finally, the core inside is revealed, ready to release the viral genome.
Reovirus core as an “mRNA factory”
Icosahedral internal core, at the base of each 12pt cortex is one part of mRNA
Has to be at base bc vertex is where RNA is extruded and copied and moved into cytoplasm
Lambda 2 (flower blooming thing) is a pentamer than forms channel so mRNA can be transcribed off of dsRNA vGenome into mRNA extruded through these turrets
Also has a capping activity enzyme in the turret, forms rna capping enzyme to put 5’ cap on. (like sending ur kid off to school LOL)
lambda 1-3
λ2: pentamers form the channels through
which mRNA is extruded; also is capping
enzyme.
λ3, μ2: RDRP subunits
λ1: RNA helicase; preps mRNA for capping
(removes terminal phosphate)
reovirus unique 4 channel structure
Start with dsRNA molecule → separated using helicase → ssRNA portion of - sense template (for mRNA copying) → enters channel #1 → as copied, exits to cytoplasm (exposed to inside of core)
In → copied → released again and cycled back to re-anneal with + sense mate
Copied mRNA as synthesized comes out channel #3, gets capped, released into cytoplasm
Channel #4 lets nucleotide triphosphates (NTPs) enter and are added on during copying.
Reovirus - replicase particles & secondary transcription
Primary transcription = primary genome copied (each 10-12 mRNA segments extruded into cytoplasm, translated via ribosome → new viral proteins)
All structural components in core are both structural and enzymatic
As proteins built up + mRNA templates = viral replication compartments
Nothing can be done right until a new core is formed around each of 12 segments → once thats done then polymerase makes -sense copy of mRNA → dsRNA genome
Now u have a core that can be used to make more mRNAs, viral proteins to amplify process
Cores are assembling outer capsid as well at the end
Most 2ndary transcription in replication compartments from cores with newly replicated dsRNA genomes
takeaway:
Reovirus replication is tightly controlled and compartmentalized: it begins with primary transcription to make viral proteins, but full genome replication (dsRNA synthesis) only happens after a new core is assembled around all 12 mRNA segments—ensuring accuracy and coordination.
reovirus pathogenesis
major cause of childhood gastroenteritis (stomach flu)
Rotavirus death usually due to dehydration bc all water gone through diarrhea
How does rotavirus cause huge fluid imbalance:
Caused by NSP4 an enterotoxin, protein, exposure of this to the gut causes fluid loss and diarrhea
NSP4 engages receptors on cells engaged in Ca signaling (important for chloride balance for inside/outside balance)
If that's disrupted → net fluid excretion instead of homeostasis
NSP4 once in cells can disrupt junctions between cells (what keeps intestinal villi in close contact) → integrity of intestine villi weakens → atrophy → hyperplasia
Villi important for nutrient absorption, of those r gone then you can't take in nutrients / fluids
Non pathogenic reoviruses can enhance development of food sensitivities—> celiac disease
Reovirus vaccine
Live attenuated vaccine
-3 re-assortants, each with a different human gene encoding a specific serotype of VP4 or VP7 (viral capsid proteins), under the backbone of a rhesus monkey strain.
Several oral, live attenuated rotavirus vaccines available
gram positive vs gram negative bacteria
Gram negative bacteria cells have an outer membrane then a thin layer of peptidoglycan then an inner membrane
Gram-positive bacteria have a thick cell wall composed of many layers of peptidoglycan and stain purple with a Gram stain.
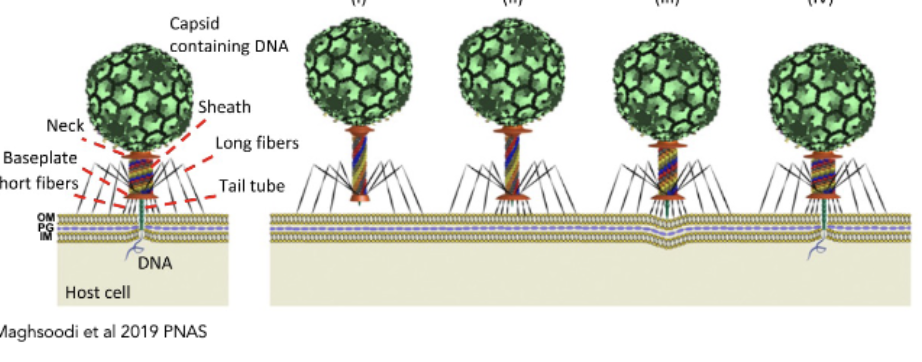
Phages delivering viral genome
pierce envelope and deliver genetic material into cell envelope
tail fibers trigger receptors → conformational changes → tail contracts, dsDNA genome into cell (like a syringe)
Transfer rate and replication rate are fast
picture is of this
unique case:
Podovirus with short tail, core proteins act like a tail, they need to be revealed. Like a spring that gets unwound when receptors are engaged. Tail and fibers like a lock and key mechanism.
bacteriophages with no tail:
they have H protein, virion binds to sugar molecules, unveils H protein which self-assembles into a nanotube that drives its way into the cell envelope and becomes a conduit for the viral genome. (like a drill)
can also use their own internal membrane as a tunnel to deliver v genome.
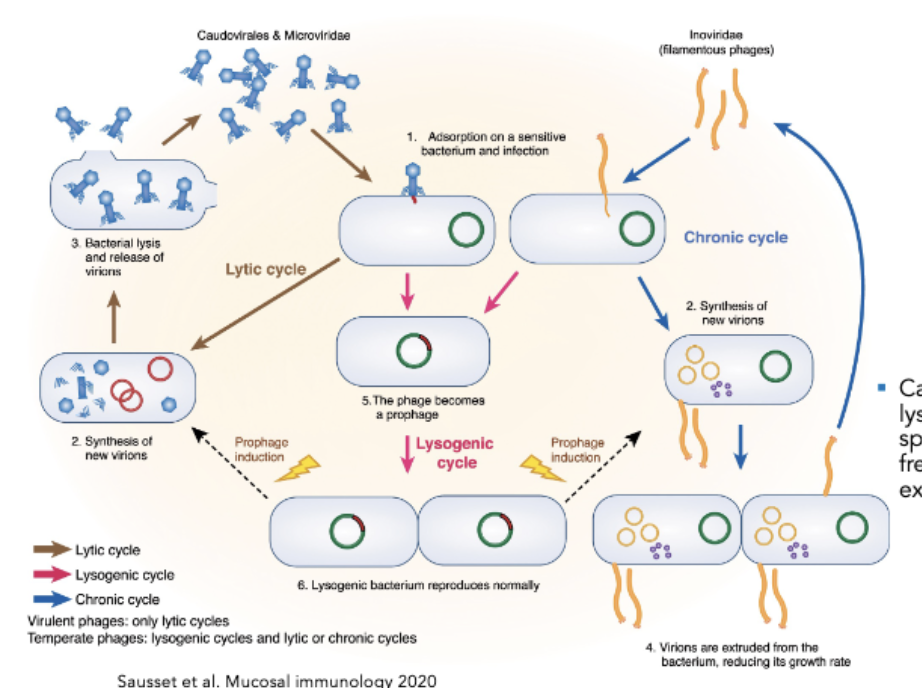
Lytic vs Lysogenic Phages
Lytic = predator-prey relationship with bacterial host, infect and kill it
Phage attaches to bacterial cell, delivers genome, takes it over to make viral copies/proteins, virions made, lyse out of cell
Lysogenic = Lysogenic phages can either enter a lytic cycle, or enter a lysogenic state
After entry of genome, genome integrated into bacterial chromosome, maintained acquiescence state, bacterium which has this prophage Integrated vgenome), goes on to divide, progeny get this DNA
Goes on forever or until the virus is queued to enter the lytic cycle
queue is usually a stressor
Note: things like filamentous phages do “non lytic release” (make their viral genome and replicate then leave with no lysis)
Phage Mediated Lysis
Need to break through cell envelope
same first steps for gram - and +
T4 complete infection cycle is 18-20 minutes
holin = inner membrane spanning protein
During cycle, holin localizes to inner membrane, phage produces endolysin (pac man), cleaves peptidoglycan but needs access to where peptidoglycan is → it accumulates when waiting
Spanin = membrane spanning protein, fuses membranes
all these proteins accumulate during infection cycle
Holin triggers large holes in inner membrane, releases endolysin into peptidoglycan → chews it up → once mesh barrier broken down, gram + lysed,
for gram - → spanin initiated to go conformational change: outer and inner membranes fuse = lysis
Pinholin = makes tiny holes LOL, holes trigger endolysin
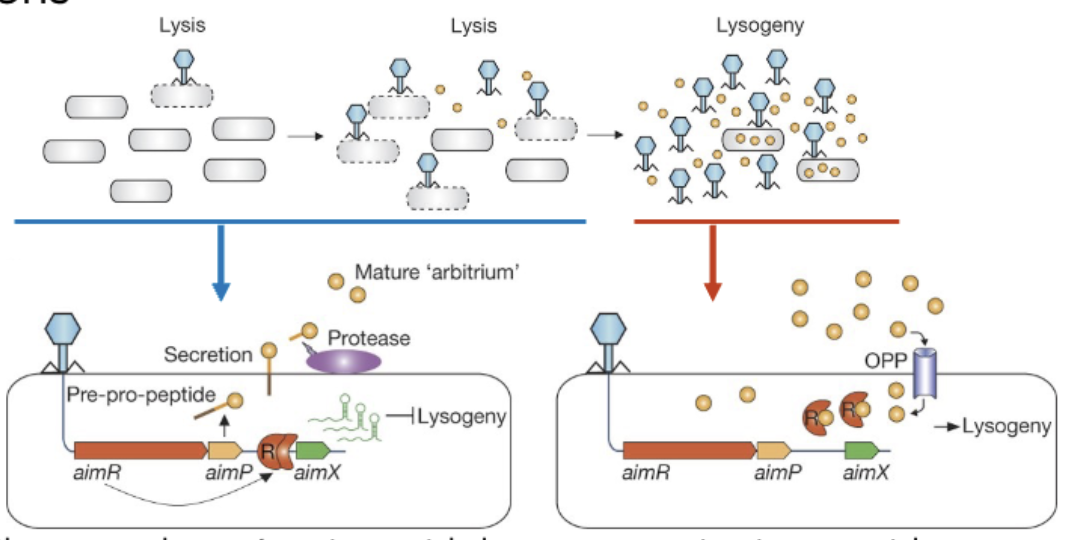
Phage-phage communication for lysis vs lysogeny
One phage kills a cell → produces small peptide → accumulates as more viruses grow
If there's a high abundance of this peptide, this environment favors lysogeny → phages know there aren't many hosts left, they can't kill off all of them → they decide to chill out LMAO
Communication = arbitrium = name of the signaling peptide
^ the peptide = AimR, it either is or isnt bound by aimP
Upon phage infection, phage comes in aimR = transcription factor which represses lysogeny = lysis chosen (left photo)
For lysogeny: TF that represses AimR gets bound to peptide (aimP) → lysogeny (right side photo)
Jumbo phage - “nucleus” and compartmentalization
GP105 builds a phage nucleus (protein shell) in center of bacterial cell.
Inside shell: DNA, replication, and transcription machinery.
Outside shell: translation occurs in cytoplasm via host ribosomes.
Shell acts like a eukaryotic nucleus: separates mRNA production from translation.
Purpose: immune evasion (blocks host enzymes), efficient genome processing.
Shell is semi-permeable: lets mRNA and small molecules through, blocks folded proteins.
Selective transport brings phage proteins (from cytoplasm) back into shell.
Filament tracks and a spindle system keep nucleus centered.
Capsids dock, load DNA from nucleus, move away to assemble tails.
Phages in human health / disease + challenges
Bacteriophages dont infect human cells —> we use natural capacity of phages to kill bacteria in our own fight against antibiotic resistance
Positive: viruses are very specific to given hosts → can target just them with phages
Challenge: narrow host specificity (reason why it prob won't become mainstream) → scientists have to find very specific phages to help treat people
bacteriophages as antibacterial agents (enzymes / process)
Endolysin is a phage enzyme that degrades peptidoglycan, causing bacterial cell lysis.
Works with holin, which creates pores in the membrane so endolysin can reach the cell wall.
Researchers can apply endolysin directly to Gram-positive bacteria (no outer membrane), killing them externally.
Doesn’t work on Gram-negative bacteria because their outer membrane blocks access.
Endolysin is produced by phages and can be species-specific, targeting only certain bacteria.
lysogenic conversion
Alteration of the phenotype of a host cell by lysogeny
Host cell becomes immune to further infection by same phage
Other phenotypic changes can also occur
ex: pf4 virion phages can be used to alter genetic makeup of a bacteria that gets in wounds and ruins everything
^ an antibiotic might make everything worse by triggering the prophage to make more virions (lytic cycle)
bacterial defense against phages
1) bacteria can change their surface molecules so phages can’t infect those cells
2) intracellular barriers to phage replication (restriction modification systems & crispr/cas) most abundant
Sequence specific systems that degrade invading virus
3) mechanistically diverse systems evolved to detect viral infection, kill cell before virus can finish replication cycle 2nd abundant
Abortive infection systems
kill urself for the tribe :(
Surface modifications (bacteria and phages) to compete w eachother
Bacteria alter expression of receptors, phages adapt to this
Bacteria can mask receptors using glycans, phages can drill through it using enzymes
Some phages have “tropism switching genetic cassettes” → they modify tail protein fibers to bind to more hosts
Done using diversity generating retroelement
Diversity generating retro element (DGE) - bacteriophages
Phages evolve to keep up with bacterial defenses by changing their surface proteins, especially the tail fibers.
Diverse population: Phage replication produces a heterogeneous population of viruses, each with different tail fibers.
Receptor binding: Phages bind to specific bacterial receptors; if bacteria change their receptors, some phages can still infect with the right tail fiber.
Stochastic process: Random chance ensures some phages with the right tail fibers can infect and replicate, even if most fail.
DGE generates this diversity in the viral population, ensuring phages can adapt to evolving bacterial defenses and continue infecting.
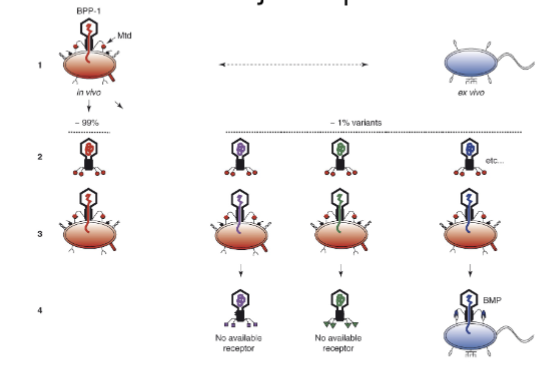
Bpp-1 DGR and mutagenic retrohoming - bacteriophages
RNA to cDNA conversion: The Bpp-1 DGR uses RNA to form cDNA through reverse transcription.
Errors during reverse transcription: Errors occur randomly at adenines, replacing the variable region of the gene in the phage.
New tail fiber tip: This results in a new C-terminal portion of the gene, leading to diverse tail fibers.
Mutagenic retrohoming: The diversity created allows the phage to maintain a diverse pool of viruses, enabling them to adapt and keep up with bacterial defenses.
same idea of random changes means bacteriophage is ready to attack / adapt to more things.
Restriction Modification systems - bacterial defense response
Bacterial defense system: Bacteria produce both a methyltransferase (MT) and a restriction endonuclease.
Marking “self”: The MT adds methyl groups to specific DNA sequences on the bacterial chromosome, marking it as "self".
Identifying invaders: Viral DNA lacks these methylation marks.
Defense action: The cognate restriction enzyme (CRE) recognizes unmodified (foreign) DNA and cleaves it, preventing viral replication.
How do some phages evade bacterial restriction enzymes?
Chemical DNA modification: Some phages modify cytosine bases in their DNA to 5-hydroxymethylcytosine (5-hmC).
Purpose: This modification prevents cleavage by bacterial cognate restriction enzymes (CREs), which normally degrade unmarked foreign DNA.
Bacterial counter-response: Bacteria evolve new enzymes that specifically recognize and cleave 5-hmC-modified DNA.
Phage’s next move: Phage adds glucosylated hydroxymethylcytosine (glu-HMC) to further shield its DNA — once again escaping host restriction.
Phages and CRISPR
CRISPR-Cas = Adaptive immune system in bacteria
Made of repeats + spacers (memory of past phage infections)
Cas genes nearby encode machinery (like Cas9)
During phage infection:
Bacteria capture snippets of phage DNA → insert into CRISPR array
Later, CRISPR array transcribed → processed into guide RNAs
Guide RNAs + Cas9 = CRISPR complex, which cleaves matching phage DNA
How phages fight back:
Mutate their genome to avoid recognition
Produce proteins that inhibit Cas9 (anti-CRISPRs)
Physically protect their genome by replicating inside protein shells
Bonus: There are CRISPR systems that target RNA, too!
basically crispr remembers phages and fights them so phages change their genome.
cGAS like pathway in bacteria + phages avoiding it
bacteria encode homologs of cGAS
virus in, queue sensed (not dsDNA bc bacteria have that), sense viral infection, produce cyclic di-nucleotides → produce effectors and kick its ass
Phages can circumvent this
using enzymes with some similarity to what infects us
Bacteria use CDNs to signal viral infection (like human cGAS-STING).
T4 phage is resistant: produces ACB, an enzyme that degrades CDNs.
No CDN = no immune signal = bacterial defense is bypassed.
Smallpox progression
10-12 day incubation period
Days 1-3 = sudden fever, prostration, nausea, vomiting
Day 5: deep lesions and pustules all over body including mouth and larynx
Scabs and bursting of lesions occurs over the next three weeks
Scab material and lesions spread virus
Smallpox - Variola Major vs Variola Minor
Variola major: hyper virulent, 30% individuals died
Variola minor: 1% lethality
Survivors are resistant to subsequent infection
WHO eradication program & vaccination strat
Humans are only known host
Vaccine highly effective against all known strains of the virus
Vaccine easy to store and maintain
Disease very easily recognized, no asymptomatic people
Can easily determine who has been vaccinated
vaccination:
Identify all individuals with disease
Trace all contacts with diseased people
Vaccinate all contacts to stop disease spread
Vaccinate everyone in vicinity of outbreaks (ring vaccination)
Advertise program to break chain of transmission
Maintain world-wide surveillance to prevent pox reintroduction
Vaccination in october (low point for smallpox)
Prevention during this period could permanently destroy a smallpox chain
General Properties of Poxviruses & challenges
Large, complex “ovoid” structures
Font have 1 set morphology → amorphous “brick like”
No defining symmetry
They're dsDNA viruses but they replicate in the cytoplasm
Smallpox challenges:
Genome replicates in a spot where its flagged as a PAMP
They can’t make use of any nuclear cellular machinery (they have to encode/package everything themselves)
Smallpox entry
Macropinocytosis: Engage receptors, but trick cell into engulfing them
Bind actin containing protrusions (filopodia), they track to the surface of the cell, once they each they induce signaling event → tricks cell into engulfment
Induces plasma membrane “blebbing”
Virus put phosphatidylserine (PS) on membrane
PS is usually facing cytosolic side of cell except when cells undergo apoptosis
^ signals to neighboring cells that it isn't a pathogen, but it's here to be engulfed
PS signaling dampens local innate immune response by telling phagocytic cells that its just a piece of an apoptotic cell
smallpox genome organization + mini nucleus
Linear dsDNA genome, no free 5’ 3’ ends
Each end is covalently closed
NOT circular
10kb inverted repeats
Genes expressed from both strands
Large genome, encodes lots of proteins
No introns in genes, prob bc virus isn't in nucleus
Vaccinia core is “mini nucleus” 41:45
Incomplete uncoating
Virus first enters, it expresses a set of genes (early genes) before it starts replicating genome, extruded from cores into cytoplasm for replication
Very similar to Reoviruses
DNA replication - smallpox
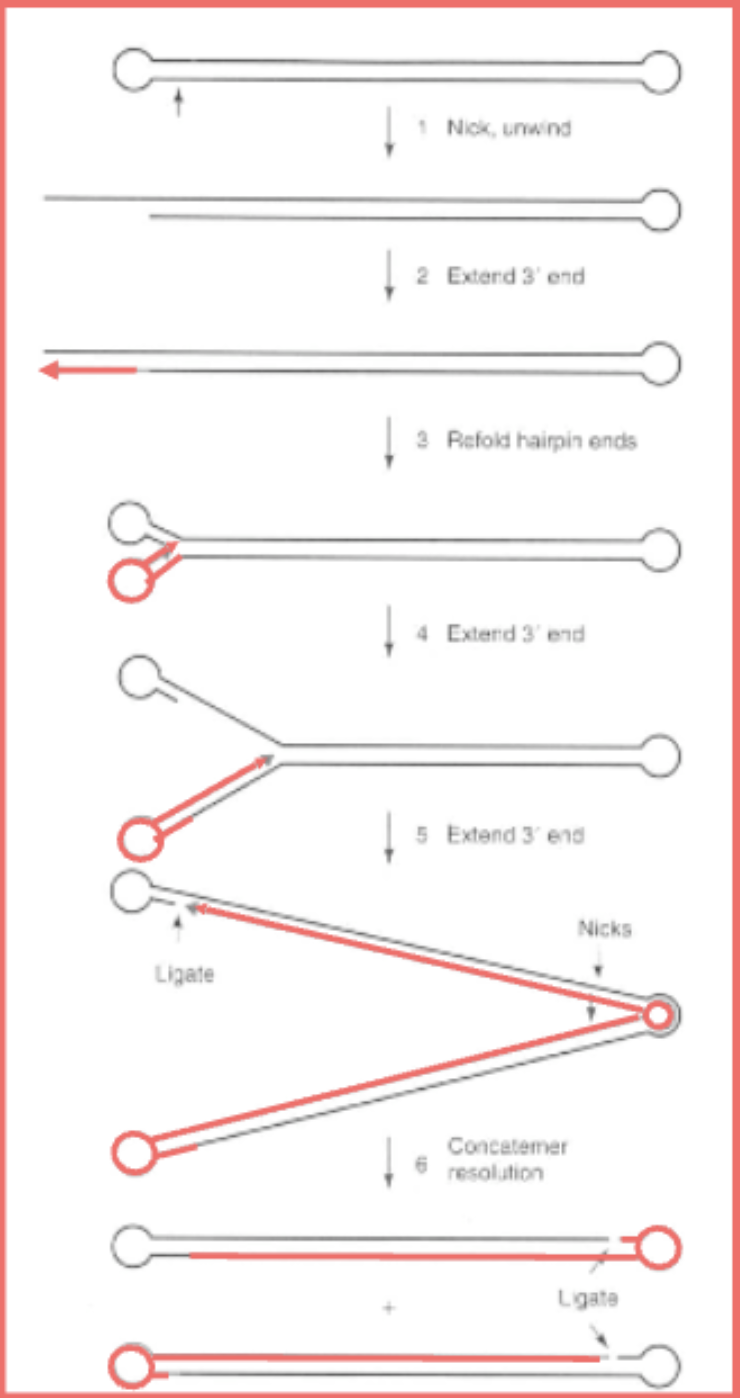
Start with linear dna with its closed ends
Virus makes nick near terminal repeats
Hairpin unwinds
Virus uses shorter end as a primer for DNA synthesis, extends till end, since its an inverted terminal repeat, it's made then fold back into a hairpin
Then it keeps going using the rest of the strand to keep going
“Strand displacement mechanism”
Nickases and ligases to resolve them into unit length structures
No free ends, terminal inverted repeats used to make long cacatomers of genome that flip back on each other to keep copying so there's never openings at the end and they can be resolved.
Vaccinia virions propelled from surface of cell by actin tails - smallpox
virus expresses 2 proteins made early on plasma membrane, these proteins induce actin tails
Actin tails are induced to be polymerized by virus
Before virus finishes replication it has tails on the surface so when
1. it finishes it can propel away
2. if there's another virus coming along, these proteins propel this virus away this cell (already infected) to a different one that isn’t infected.
SV40 (polio, hit and run tumorigenesis)
SV40 was a contaminant of early 1960’s polio vaccines
There ended up being no risk, bc it doesn’t cause cancer in humans
“Hit and run model of tumorigenesis” = tumor inducing agent plays a casual role in initiation but not maintenance of the tumor
sv40 strength and uncoating
SV40 is super strong bc VP1 protein (dont need to know name?), forms a pentamer which becomes extensively cross linked to itself via disulfide bond, like molecular chain mail
How do you reverse this to get the viral genome out?
SV40 VP1 protein forms a pentameric capsid crosslinked by disulfide bonds — very stable, like molecular chainmail.
To escape, SV40 must uncoat its genome by reversing these crosslinks.
Enters the cell via lipid raft microdomains (not typical receptor-mediated endocytosis) → this sends the virus to the ER.
In the ER, protein isomerases break disulfide bonds, loosening the capsid.
SV40 mimics a misfolded protein, triggering the ERAD (ER-associated degradation) pathway, which exports it to the cytoplasm.
In the cytoplasm, the virus uses the microtubule network to travel to the nucleus, where infection proceeds.
vp2 and vp3 have DNA binding domains and nuclear transport signals, potentially used in delivering genome to the nucleus.
polyomavirus genome structure and organization
Super tiny genome, not all DNA viruses are large
Circular genome, packed with nucleosomes (cellular histones) from cell
Don't make your own DNA organizing protein, steal it!
All DNA viruses have kinetic organization of gene expression (early genes = transcribed before virus replicated, late = genes transcripted after virus replicates)
Early are needed for cell setup (machinery cell encodes itself)
Late genes (structural capsid components)
Early and late promoters
T = tumor antigen (viral antigen)
VP2 & 3 have nuclear localization signal
bidirectional DNA replication - polyomaviruses
Bidirectional DNA Replication in Polyomaviruses
Polyomaviruses (like SV40) have a circular double-stranded DNA genome.
Replication starts at a single origin of replication (Ori).
From the origin, DNA replication proceeds in both directions — this is called bidirectional replication.
This process creates a structure that looks like the Greek letter theta (θ) — often called a theta replication intermediate.
5’ end problem
DNA polymerase can’t fill in the 5′ end after RNA primers are removed on linear DNA, leading to loss of sequence.
But circular DNA avoids the 5′ end problem, allowing complete replication without losing genetic material.
regulation of S phase & how viruses hijack it
DNA virus can stay small bc they steal DNA replication from host cell
human regulation:
Viruses push cells into S phase to access DNA replication machinery, often by inactivating Rb to free E2F, which turns on replication genes.
(e2f in cytoplasm)
Normally, Rb holds E2F inactive; CDKs phosphorylate Rb during proper cell cycle entry.
p53 monitors for inappropriate replication and blocks CDKs or triggers apoptosis to prevent cancer — viruses often try to disable p53 to avoid this.
P53 can block cyclin activity outside of its job, if that doesn’t happen it says apoptosis time. Kys. = no cancer
Virus overcoming:
SV40 gets all this done with T bc it has rb binding domain, also has p53 binding domain to inactivate it
Most of these don’t cause cancer bc cell dies
Large T comes in, it inactivates RB and p53 → leads to S phase (involves turning on CDKs, they also phosphorylate T → signal to T that S phase successfully was turned on, turn ur activity to viral DNA replication/binding, triggers binding to DNA origin of replication and start replication
Polyomavirus - Large T antigen
Inactivates Rb (Retinoblastoma protein)
LT binds to Rb → releases E2F → pushes cell into S phase → host cell starts making DNA replication machinery, which the virus needs.
2. Inactivates p53
LT binds and inhibits p53, preventing apoptosis → infected cell stays alive long enough to make more viruses.
3. Binds the Viral Origin of Replication (ORI)
LT recognizes and binds specific sequences at the viral ORI (origin of replication).
hexamer binds it?
Binding here is essential to start viral DNA replication.
4. ATPase/Helicase Activity → "Melts" dsDNA
LT has ATPase activity (it uses ATP for energy) and acts like a helicase.
This allows LT to unwind/melt the double-stranded viral DNA at the origin → creates a single-stranded region where host DNA polymerase can start replicating the viral genome.
Papillomaviruses: general
huge cause of cervical cancer
Small, dsDNA genome
Naked capsid
Circular dsDNA genome (like polyoma)
DNA is packaged using cellular histones as DNA organizing material (don't encode their own dna binding proteins)
Huge diversity, can infect vertebrates
Stays within species
Infect epithelia, some have a preference for skin, some have a present for mucosal epithelia
There are some cell and tissue specific viruses, some really only want to stay in one part of the body (bottom of foot wart for ex)
Can be Low risk/benign and also cancerous
Papillomavirus genome structure & organization
10 ORFs
E = early (DNA replication, viral life cycle, dna rep prep)
L = late (capsid proteins, late stuff.)
Control region (promoters, transcriptional regulatory elements)
basal lamina - Papillomavirus
Infects basal cells via microabrasions
basal cells are stem-like cells that divide and give rise to differentiated skin cells.
Early proteins E1/E2 replicate DNA & repress E6/E7
As cells differentiate, E6/E7 turn on to keep cells in S phase
E6 and E7 slow down differentiation and keep the cells in S phase, so host DNA replication machinery stays active (benefiting the virus).
In upper layers, late genes (capsid) expressed → virus assembles
Virus sheds as cells die → avoids immune detection
Warts persist due to continuous infection of basal cells
E6 and E7 - Papillomavirus
Differentiated cells no longer divide so virus must induce S phase to hijack DNA replication factors
HPV E7 binds retinoblastoma (RB) to displace E2F so E2F goes to nucleus and turns on DNA synthesis genes
E7 also promotes CDK2 which phosphorylates RB further dissociating it from E2F
Machinery on, if E2F released before planned it can get detected by p53 which will lead the cell to apoptosis
Every virus that needs DNA replication needs E2F and also has to deal with p53 activation
E6 targets p53 in papilloma → it induces proteolysis so p53 is degraded
Triggers E6ap ubiquitin ligase, transfers ubiquitin to p53 → degraded

3 phases of DNA replication. Papilloma
1) establishment phase: viral genome amplified to 100 copies in undifferentiated basal cells
2) maintenance phase: viral genome replicates only once per cell cycle as cell divides, 1 daughter cell remains, 1 differentiates. Gives cell an initial pool
3) productive phase: high levels of viralDNA per cell (1000 per cell) in terminally differentiated cells
E1 and E2 origin binding proteins, induce binding of DNA
Dna linearization and cancer - papilloma
Rarely, the viral DNA linearizes randomly & integrates into host cell genome.
When the break occurs in E2 region, removal of the E2-mediated repression of E6/E7 predisposes cells to transformation (dysplasia): tumors express E6/E7 but usually not E1/E2!
Adenovirus Genome
Medium sized
Linear dsDNA
Has Inverted terminal repeat sequences, used for DNA replication
Covalently linked protein (terminal protein) = protein primer, used to avoid losing genetic info at extreme ends
Early and late genes as well
(see photo)
E1A and E1B genes = tumor antigens, coordinate entry into S phase, regulate transcription of other viral genes
E3 = stealth region of genome, packed with genes that counteract adaptive and innate immune system
adenovirus ca be used in tumor cell killing and cancer immunotherapy

adenovirus attachment and internalization
1. Fiber protein binds to 1° receptors (CAR, MHC I, etc; widespread).
2. Penton base binds integrins (more selective), promoting virus uptake by receptor-mediated endocytosis. Pentons lost.
3. Entry into cytosol is pH dependent. Viral protease cleaves/ activates membrane-permeablizing protein present in virion.
4. Microtubules carry partially uncoated nucleocapsid to nuclear pores, where they bind cellular proteins that promote capsid disassembly and import of viral DNA into the nucleus.
E1A & E3 - adenoviruses
first protein to be made after virus entry and is required for
activating viral transcription and promoting viral DNA replication
no intrinsic DNA binding or enzymatic actvities, it coordinates dozens of celluar proteins
32 primary interactions with cellular hub
E1A is intrinsically disordered, and samples various conformations to maximize the number of interactions
E1A have short linear interaction motifs (slims) = short stretch of amino acids that do adopt a particular structure, these are how it forms a series of connections and un-form them when they're done
Has conserved regions too, same across all E1A proteins → these must be important & specific
2 of these regions are similar to HPV’s E7 (binds RB), and SV40 (binds RB and p53)
Nucleates formation of the RNAPII preinitiation complex by binding promoter targeting transcription factors and co-activators. (kick off gene expression)
By freeing E2F, E1A activates transcription from a variety of genes containing E2F-binding sites in their promoters, including the adenovirus E2 early promoter
• Binds tumor suppressor Rb to induce S phase, viral & cellular transcription.
e3 = The E3 region encodes proteins that
(1) inhibit immune cell killing pathways and
(2) prevent MHC molecules from reaching the cell surface, shielding infected cells from detection by the adaptive immune system.
adenovirus inactivating Rb and p53
E1b and e4orf6 use cellular ubiquitin ligases to tag p53 and send to proteasome for degradation
E1B 55k, binds p53 directly and inhibits activity
Bax = protein on mitochondria which does cell death when activated
E1B 19K Bax,
Mimics Bcl-2 protein.
Inhibits Bax at mitochondria → prevents cell death.
Methylation machinery can also be required to stop activation
adenovirus genome replication
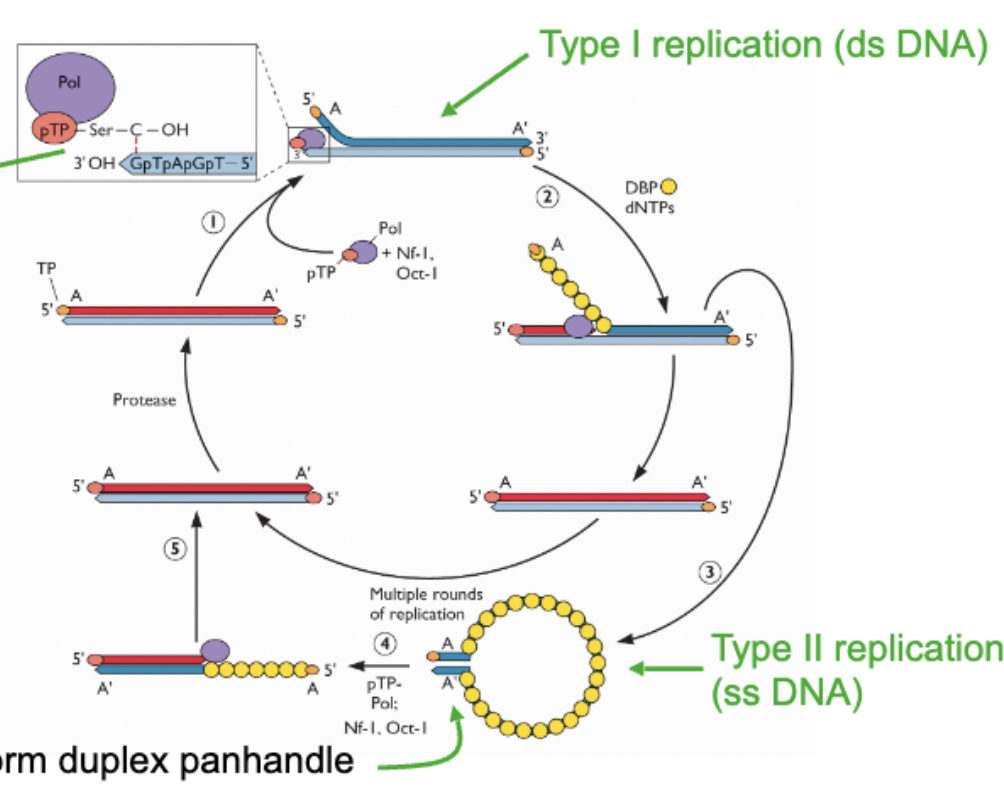
Start with dsDNA genome → start using terminal protein which is connected to DCMP nucleotide → position protein primer at extreme end through base pairing between C residue and G residue at end of genome → polymerase comes and primes off of this , copies genome, as copying happening strand displacement happening to strand not being copied → at end an entire strand is displaced and replaced → this is type 1 replication
After this ^
The ssDNA that got released coated with ssDNA binding protein so it doesn’t fold back → inv terminal repeats allow it to form panhandle structure which looks like dsDNA end → this region is protein primed to start replication in the same way as type 1 → priming starts, replication starts → boom two dsDNA molecules → type 2
Important: Protein priming replication, 2 stages of DNA replication
Herpesvirus (general)
Large, enveloped dsDNA viruses,
have cycles of latency and active / lytic replication
really good at spreading through human population
Once you get herpes you are infected for life
can be asymptomatic
Viruses can be spread with or without visible lesions, through saliva, mucous membranes, mother to child
Super bad for immunocompromised people
For most of us we have a balance where we have “co-evolved” with them
Viruses are species specific, they get specialized for the host they infect
Once in nucleus → can circularize: form episome = DNA not integrated but tethered to host chromosome, genome preserved till it gets a signal to replicate (silent state) or active
3 families; Alpha, beta, and gamma
Amorphous region between capsid and envelope = tegument
Packed with thousands of viral/cellular protein/RNAs delivered to host cell the second the virus fuses with host cell
Herpes encode their own DNA polymerases
Herpes entry machinery
Has a ton of viral proteins that all regulate fusion and receptor binding they have to collaborate with each other in ways not fully understood
G = glycoprotein shown on picture slide 6 (important in red box)
Not talking about accessory factors
Main set:
gD = receptor binding undergoes conf change allowing it to bind gH and gL
gH & gL complex = acts as an intermediary between gD and gB, transducing signals from the interaction of gD with receptors.
gB = gets put into fusogenic state by gL and gH, primary membrane fusion protein, physically fuses the viral and cellular membranes, so virus can enter host cell
tegument proteins (virus entry)
VHS, VP16, PML bodies, PP71, IE1
Tegument factors are setting up favorable environment in host cell
Example of tegument proteins:
VHS = virion host shutoff factor, a nuclease, cuts up RNA
Frees up ribosomes so there's no competition, creates an overall immune evasion phenotype, nuclease degrades cellular RNA = kills antiviral encoding transcripts = dampens innate immune responses
VP16: transcription activator, kick starts production of immediate early genes which turn on early genes
Some tegument proteins prevent permanent silencing of viral genomes by targeting PML bodies
PML bodies (host) = substructures in nucleus, contain epigenetic modifiers, silence DNA by putting restrictive methylation marks to condense it, makes it inactive = intrinsic restriction
Evading PML bodies: pp71 tegument protein goes into nucleus and causes dissociation of 2 of epigenetic modifiers present in PML bodies
ie1 = causes dispersal of PML to prevent repressive activity.
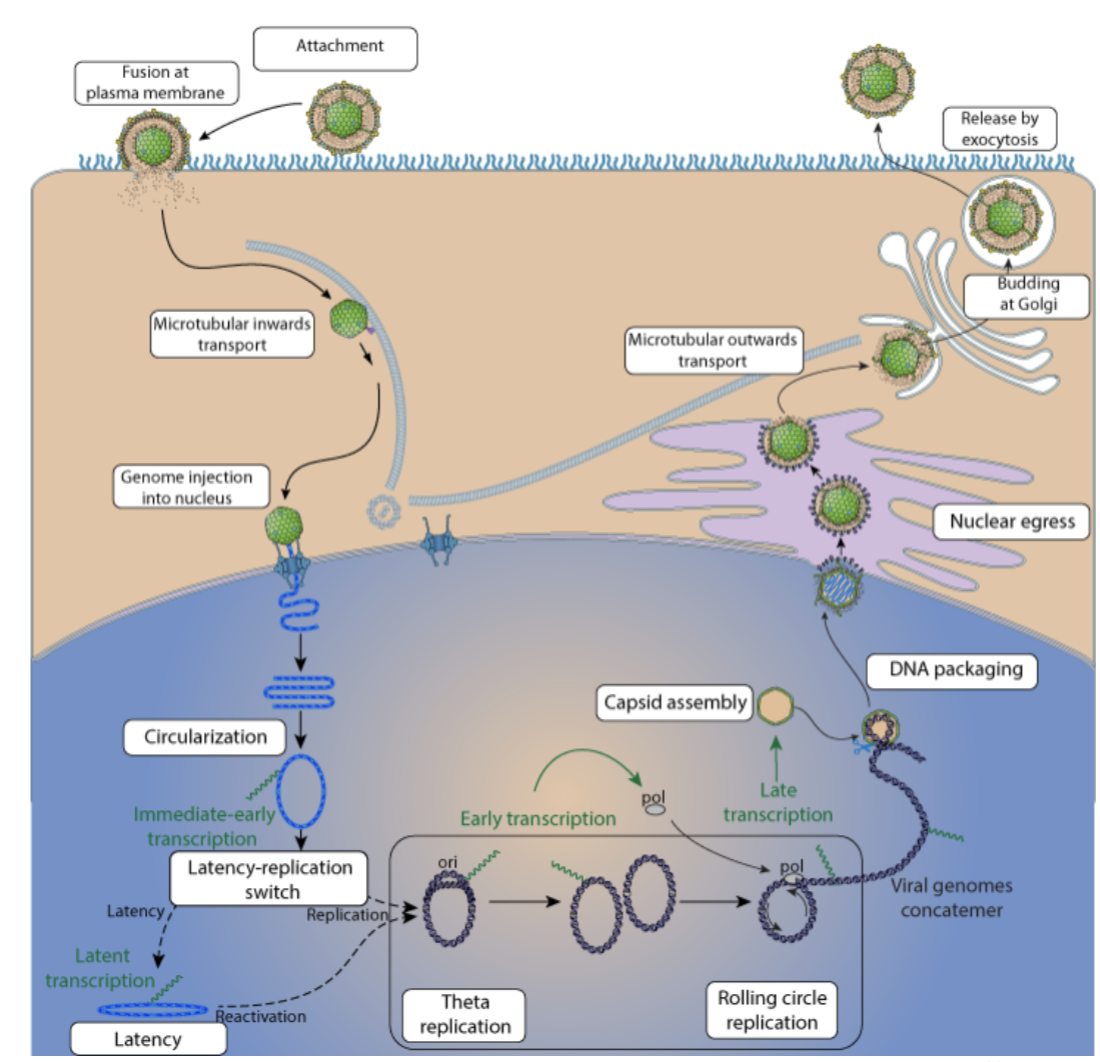
Herpes Viral Replication
Blocks the cell in G1 = not known why
DNA replication strat is rolling circle mechanism:
nick made, DNA synthesis from 3’OH end (discontinuous), keeps going, resulting in 2 double stranded DNA circles?
Virally encoded proteins for genome replication:
✓DNA polymerase (2 subunit)
does NOT encode rna poly
late gene trascription coordinated by a protein mimicking tata binding protein
The virus makes its own version of TBP (a TBP mimic).
This mimic can directly bind to viral late gene promoters and bring in RNAPII right away.
So, it bypasses the need for the rest of the host’s transcription machinery.
more “Streamlined”
Herpes —> packaging viral genome
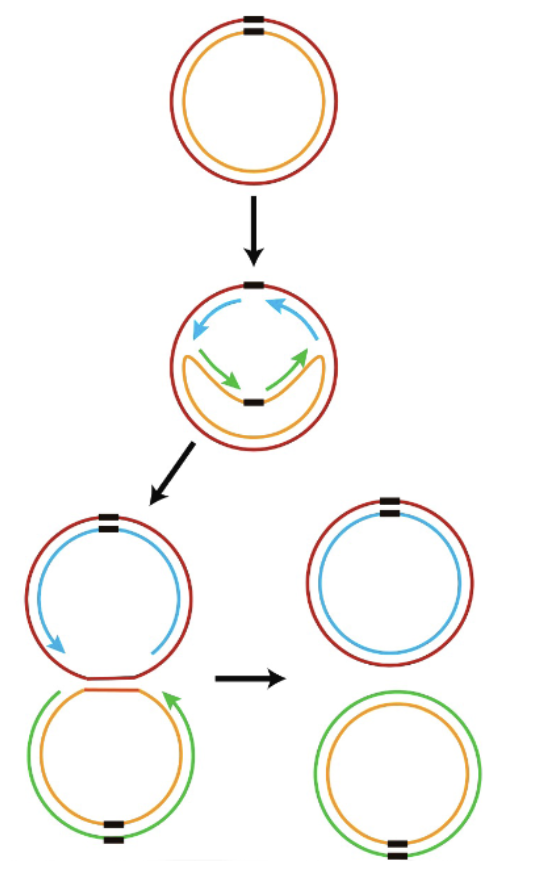
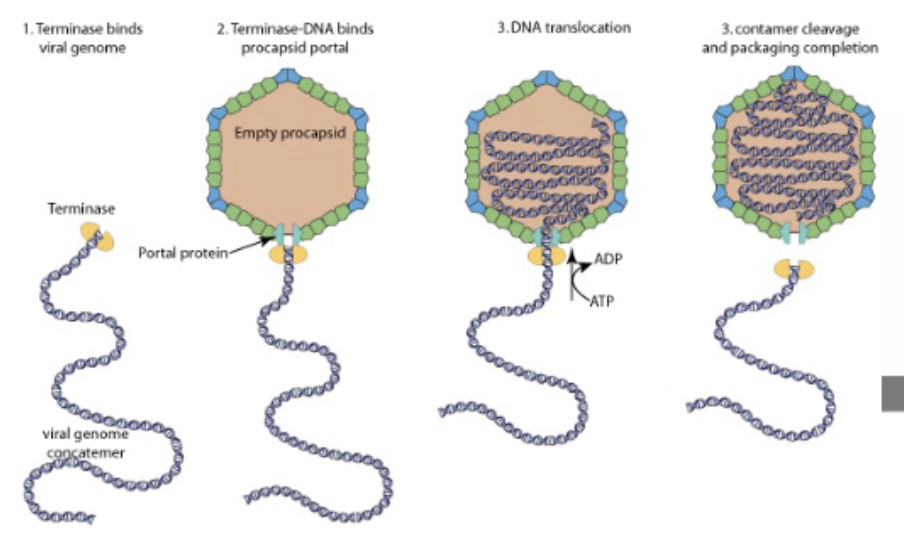
viral genome concatemer + procapsid + terminase
terminase binds inverted terminal repeats on viral DNA, takes it to portal on icosahedron (empty procapsid), ATP dependent process —> translocates the DNA, feeds it in until next terminal repeat, nuclease activity cleaves it, portal sealed, DNA packaged icosahedron matured
Herpes exiting the cell
Has to disrupt nuclear lamina to escape into the cytoplasm to capture the tegument
Buds into the nuclear membrane on the nuclear side, Buds out on cytosolic side
Catches tegument factors, buds into transgolgi network, gains its lipid envelope and is they exocysed out of cell
The nuclear membrane is hard to break through, but herpesvirus hijacks CDK activity, just like in mitosis, to weaken it so the virus can escape the nucleus.
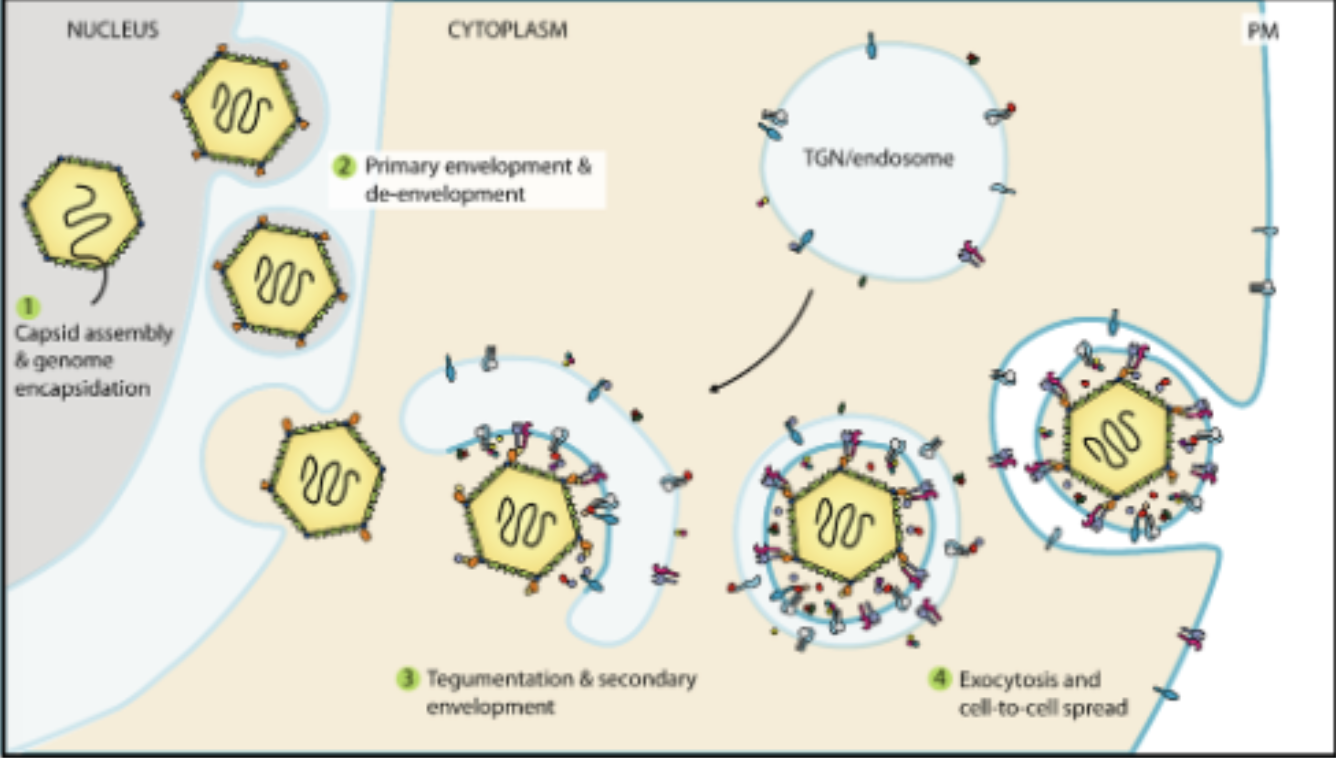
herpes replication cycle (overview)
attachment
fusion
microtubular inwards transport
genome injection into nucleus
circularization
latency replication switch
latency
OR
theta replication
Rolling circle replication
Capsid assembly
DNA packaging
nuclear egress
microtubular transport outwards
budding at golgi
release by exocytosis
Herpesvirus latency (general)
Gamma is the only family that is oncogenic (causes cancer)
Alphas establish latency in neurons:
it traffics along sensory neurons to deposit genome in a quiescent state, no viral proteins expressed.
They hang out in non dividing cells
When the host is stressed → triggers virus to reactivate, travels back down axon, releases viral particles, causes local lytic reactivation.
Cold sores: those are where herpes is latent in
Beta HV: quasi latency state, hang out in monocytes and macrophages
Gamma HV: humans pathogens (epstein barr, sarcoma) establish latency in your B-cells, oncogenic. Most of us infected with EBV
B cell differentiation
Virus infects naive B cell
shows version of antibody it will make on cell surface, if it recognizes smt with antibody → becomes active
Virus drives naive b cells to go to germinal center and proliferate as if they did find an antigen → creates a stable long lasting reservoir for the viruses (bc b cell was made into a memory b cell)
From infection to proliferation:
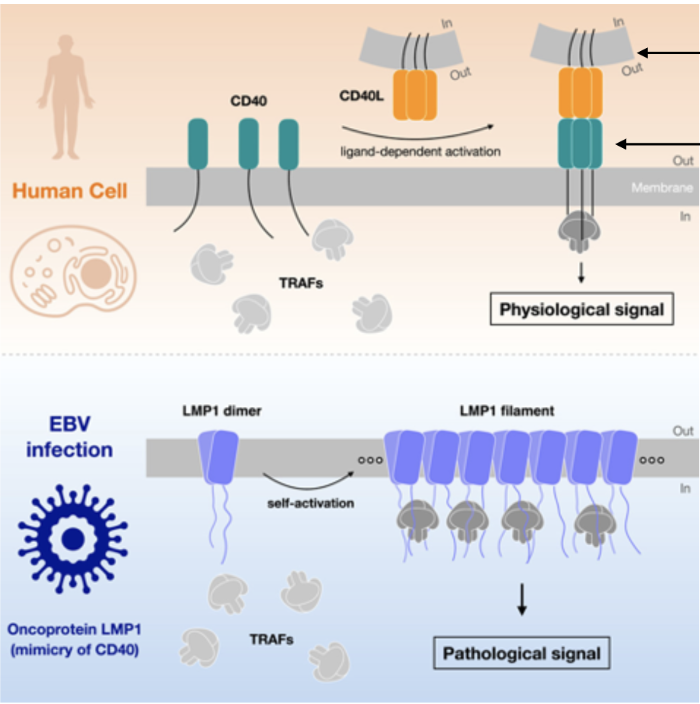
EBV infects a B cell.
LMP1 (a viral protein) gets expressed in the B cell membrane.
LMP1 mimics CD40, a normal receptor that usually requires T cell interaction (via CD40L).
But LMP1 doesn’t need CD40L — it's always “on.”
LMP1 recruits TRAF proteins (TNF receptor-associated factors).
Result: B cell thinks it’s being told by a T cell to grow, divide, and stay alive.
This allows the virus to maintain latency and expand infected B cells without immune activation.
Move out of germinal center and into memory cell
Memory cell divides, viral genome divides with it so it doesn't get diluted out
Doesn’t express viral proteins → undetected
Back into lytic cycle: when b cell gets differentiated into plasma cell which happens bc b cell comes in contact with its matching antigen
how does cancer happen - herpes
Virus needs to drive cell progression in absence of approporate signals
latency program encodes viral proteins with growth stimulating potential
Oncogenes
Activation of the ‘growth program’ by EBV during B cell activation
poses a cancer risk, but is usually counteracted by the virus and the
immune system
main note: lots of latency genes have oncogenic activity
Epstein–Barr Virus (EBV) and Multiple Sclerosis (MS)
MS = Autoimmune disease, Eats away myelin sheath on nerves → causes tons of symptoms
EBV is main / only driver of MS
This is because of an autoreactive antibody produced during initial burst
Some antibodies against EBNA-1 end up attacking GlialCAM which is what coats the neurons/myelin sheath bc it looks the same
These antibodies end up cross reacting with myelin sheath protein and attack it.
Normally NK cells and T cells kill reactivated EBV infected plasma cells that produce autoreactive antibodies
Sometimes, individuals infected with EBV have immune cells that present a weird form of MHC-1 (HLA-E)
This form (HLA-E) can inhibit NK cell activity when loaded with another peptide from EVB protein LMB-1 (CD40 mimic)
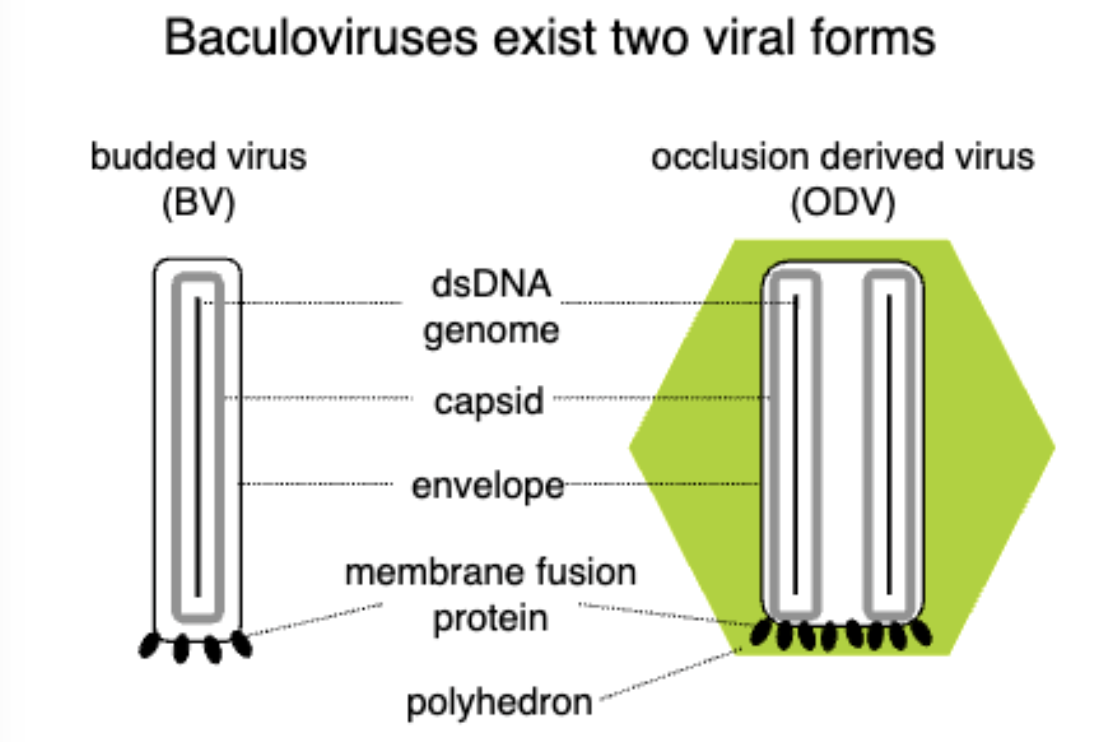
Baculovirus Biology
Used to kill insects, environmentally friendly pesticide, mammalian cell transduction vectors.
liquifies insects…
2 forms:
Occlusion is released when insect is liquified, this is the one that hits other insects
have this extra outer layer for extracellular environment, gets degraded to become budded.
Budded virus is cell to cell transmission
Budded form buds off from plasma membrane, occlusion bodies carry virions in a crystallized structure which contain a lot of nucleocapsids.
Note:
budded viruses made during early replication, occlusion viruses made during late replication.
Baculovirus Pathology
Infects insects
Insects eat crystals, gets into the gut of the insect, infect epithelial cells in gut of insects, either replicate in epithelial cells (nucleocapsid makes more nucleocapsids) or they pass through epithelial cells into deeper tissues of insect
Budded form and occlusion forms made
(complex life cycle slide ^)
Actin in baculoviruses
Actin facilitates viral movement: Actin filaments help the baculovirus virions move within the host cell by using actin polymerization for intracellular transport.
Virus egress: Actin is involved in the cell-to-cell spread of the virus, helping newly formed virions move toward the cell membrane for budding or cell lysis.
Cell-to-cell transmission: During infection, actin-mediated movement helps viral particles move through the cytoskeleton and spread between neighboring cells, ensuring continued infection.