Arrhythmia MDRs + Tissue Oxygen Monitoring SOTA + Cornell Cardiovascular Videos
1/158
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
159 Terms
What has fast response action potentials and what has slow response action potentials?
Atrial and ventricular myocytes have a fast response action potential with 5 distinct phases
Sinus and Atrioventricular nodes have a slow response action potential
Phase 0 of Fast Action Potentials
Depolarization phase
Rapid upstroke due to opening of fast sodium channels and sodium influx
+ ions going into the cell
Phase 1 Fast Action Potential
Beginning of repolarization due to transient K efflux vi IKto channels
+ ions out of the cell
Phase 2 of Fast Action Potential
Early repolarization phase
The plateau is caused by sustained slow calcium influx via ICa(L)
K+ still going out so maintaining
Phase 3 of Fast Action Potential
Late repolarization phase
The downward slope is due to the cessation of calcium influx as the potassium efflux continues
+ ions going out, none going in
Phase 4 of Fast Action Potential
Resting phase
Maximum resting membrane potential (RMP) is approximately -90 mV primarily due to Na+/K+ ATPase pump (K+ in, Na+ out)
Entire Fast Action Potential

Phase 0 and 2 Slow Action Potential
No phase 1
Calcium influx (ICa-L) via voltage-gated L-type Ca channels (absent Nav1.5 channels)
Phase 3 Slow Action Potential
Efflux of potassium
Phase 4 Slow Action Potential
No stable phase 4 (not “RESTING”) due to depolarizing currents
Influx of Na+ (If, via HCN channels, aka the “funny” current)
Influx of Ca2+ (ICaT via T-type Ca and ICaL via L-type Ca2+ channels)
Entire Slow Action Potential

Excitability
Ability of a cell to generate an action potential in response to a stimulus depends on availability of Na+ channels to open in response
Automaticity
Ability of a cell to spontaneously generate an action potential
The SA node is the dominant pacemaker due to its faster rate
Classically other subsidiary pacemakers include the AV node, His-Purkinje system
The subsidiary pacemakers are normally inhibited by the faster rate of the sinus node - called overdrive suppression
Refractory Period
Time when myocytes are non-excitable
The refractoriness of the action potential is primarily due to inactivation of Na+ channels soon after onset of the action potential
Total Refractory Period
Effective refractory period + relative refractory period
Effective Refractory Period
Absolute refractoriness - phase 0 until halfway through phase 3 (~-50 mV membrane potential)
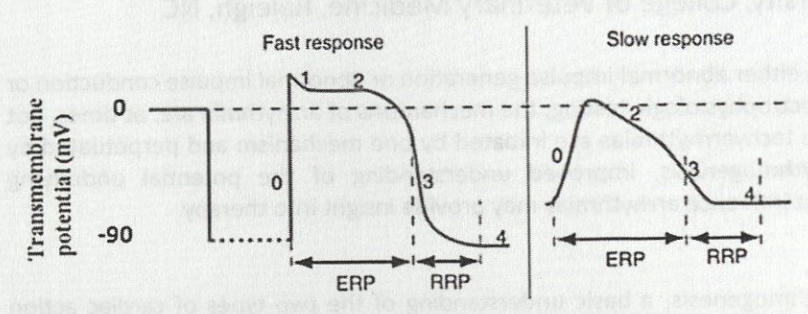
Relative Refractory Period
Myocytes can respond to very intense stimuli - from end of effective refractory period to end action potential; that is from last portion of phase 3 to initial portion of T wave → during which Na+ channels progressively re-activate (-50-90 mV membrane potential)
Conduction
Property of cell to propagate impulse from one cell to another depends on: slope and amplitude of phase 0, diameter of cells, number of intercalated discs, and connexins in gap junctions
Increased slope and lower resting membrane potential = higher conduction
Myocardial Heterogeneity of Action Potential
Action potential duration and shape differs from endocardium to epicardium
Epicardial myocytes action potential have prominent phase 1 and doming shape whereas mid-myocardium action potential have longer action potential
Differences in duration and morphology of action potentials reflect different expressions of Ito and Ik potassium channels
These differences in action potential generate transmural electrical gradients during repolarization leading to electrical heterogeneity that can serve as substrate for arrhythmias, such as re-entry
Atrio-ventricular and Ventriculo-atrial Conduction
Concealed conduction describes changes to the surface ECG that suggest altered AV nodal conduction caused by an earlier event hidden on the surface ECG
e.g. an unexplained PR prolongation or blocked P wave due to either partial or complete refractoriness of the AV node
Antegrade conduction through the AV node is also affected by concealed conduction after APC, atrial tachycardia, atrial flutter, atrial fibrillation
Concealed conduction may result from retrograde ventriculo-atrial conduction of VPC, escape rhythm, junctional rhythm, pacemaker beats
It may also occur via accessory pathway, in which atria are activated sequentially
Causes of Cardiac Arrhythmias Due to Abnormal Impulse Generation
Enhanced or suppressed automaticity
Abnormal automaticity
Triggered activity
Enhanced or Suppressed Automaticity
Changes in the spontaneously depolarizing cells of the sinus node, the atrioventricular junction, and cells of the His-Purkinje system
Ionic basis of enhanced automaticity (e.g sinus tachycardia) is explained by a net gain of intracellular positive charge during diastole causing a a steepening of phase 4 depolarization
The sinus nodal discharge rate keeps dominance over latent pacemaker sites because it depolarizes more rapidly by overdrive suppression
Can cause prolonged suppression of normal pacemakers in proportion to the duration and rate of stimulation by a more rapidly discharging pacemaker
Mechanism is related to active Na+ extrusion during the more rapid rate that maintains the diastolic depolarization of the latent pacemakers at a level more negative than the threshold potential for automatic discharge
Abnormal Automaticity
Refers to when the dominant pacemaker shifts to a site other than the sinus node such as atrial, junctional, or ventricular myocytes
These cells gain the ability to generate an ectopic impulse due to ischemic injury or electrolyte disturbances
May cause premature beats, atrial tachycardia, accelerated idioventricular rhythm, and ventricular tachycardia
Triggered Activity
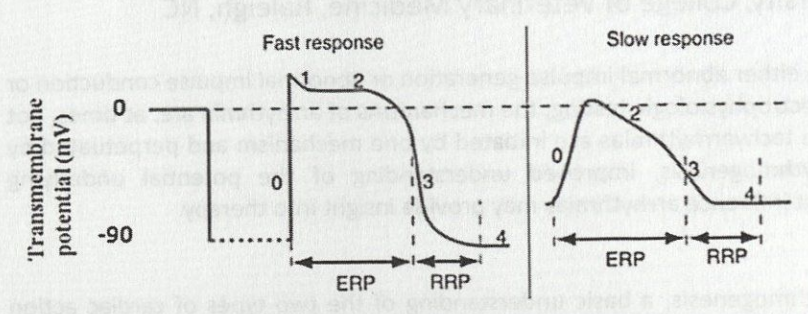
Caused by membrane potential oscillations, called after-depolarizations, which occur during or immediately after an action potential
Early after-depolarizations (EAD) - phase 2 or 3 or the action potential
Delayed after-depolarizations (DAD) - phase 4 of the action potential
After-depolarizations cause arrhythmias when the membrane potential oscillations reach the threshold potential and starts a new AP
Not all after-depolarizations reach threshold potential, but if they do, they can trigger another depolarization and be self-perpetuating
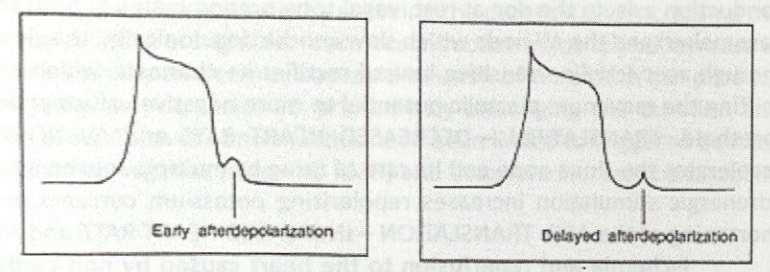
Predisposing Factors for Early After-Depolarizations
Long cycles (slow HRs)
Hypokalemia
Hypocalcemia
Hypomagnesemia
Cardiac and non-cardiac drugs that prolong the QT interval
How to treat early after-depolarizations?
Magnesium supplementation
What causes delayed after-depolarizations?
Caused by a transient inward current that is small or absent in normal conditions
When intracellular calcium overload occurs (adrenergic stimulation, hypercalcemia, prolonged APDs, rapid repetitive stimulation, digitalis toxicity), increased Ca2+ stimulates both Cl- currents and Na/Ca exchanger, resulting in transient inward currents and after-depolarizations
Delayed after-depolarizations thought to be the primary mechanism of arrhythmias in the failing myocardium and digoxin toxicity
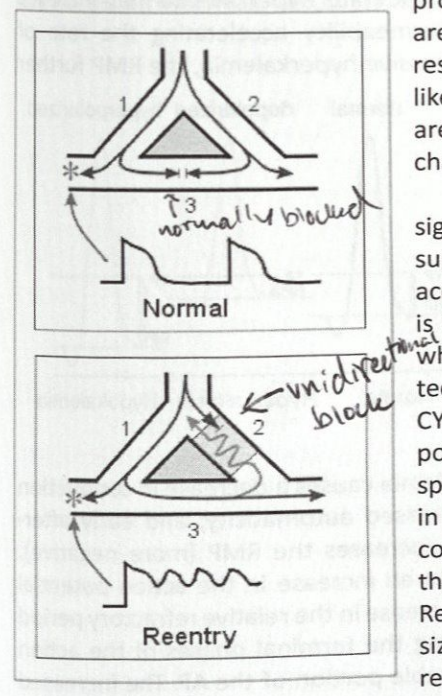
Reentry
Underlying mechanism for most ventricular tachycardias and many supraventricular tachyarrhythmias such as atrial fibrillation, atrial flutter, AV accessory pathway tachycardia
Require a circuit with a fast and a slow pathway
An ectopic discharge is blocked unidirectionally at the zone of slow conduction
The impulse travels through the normal tissue and slowly penetrates the zone of slow conduction “backwards” and creates the reentry tachycadia
Therapy for Reentrant Tachycardia
Break the cycle
Done pharmacologically by prolonging the action potential duration (increasing refractoriness) or increasing conduction impulse speed
How does body temperature affect heart rate?
Increased body temperature increases heart rate by increasing the slope of phase 4 of the action potential
Low body temperature will decrease the heart rate by affecting the rate of discharge from the sinus node
Where are the effects of hyperkalemia most and least pronounced?
Effect is more pronounced in atrial than ventricular myocytes
Spontaneously depolarizing tissue (specifically the sinus node) least sensitive
Effects of Hyperkalemia on Cardiac Rhythm
As extracellular potassium levels increase, the resting membrane potential becomes less negative
Mild to moderate hyperkalemia may increase excitability and conduction velocity by its effect of increased membrane permeability accelerating the rate of repolarization and shortening the action potential duration
With progressive hyperkalemia, the resting membrane potential further depolarizes (becomes less negative) decreasing Na channel availability
First happens in the atria, leading to a state of constant depolarization and loss of excitability
Loss of P wave, widening of QRS, and slower than expected HR on ECG are likely first indications of a life-threatening hyperkalemia
Atrial standstill or sinoventricular rhythm
Severe hyperkalemia causes a slowing of conduction velocity eventually to the point of propagation failure and inexcitability of both atria and ventricles → asystole or ventricular fibrillation
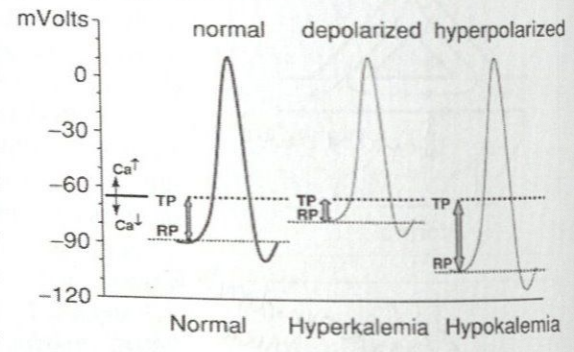
Effects of Hypokalemia on Cardiac Rhythm
Causes decrease in conduction velocity in cardiac tissue, prolongation of the relative refractory period, increased automaticity, and early afterdepolarizations
Increases the resting membrane potential (more negative)
Results in increased QT interval
Increase in the relative refractory period and a decrease in the difference of the resting membrane potential from the threshold potential during terminal phases of the action potential causes cardiac cells to have increased excitability during a considerable portion of the action potential
Causes ventricular ectopic complexes, supraventricular ectopic complexes, and AV conduction disturbances
Effects of Hypercalcemia on the Cardiac Rhythm
Shortens AP duration primarily by reudcing duration and increasing the amplitude of phase 2
Arrhythmias rare but rapid infusion of IV calcium can cause bradycardia, extrasystoles, and AV block
Effects of Hypocalcemia on the Cardiac Rhythm
Can increase atrial premature depolarization by prolonging the duration and amplitude of phase 2
Can see prolonged QT interval
Makes threshold potential more negative so increases excitability
What are the most serious and concerning arrhythmias in dogs?
Sustained and non-sustained ventricular tachycardia, supraventricular tachycardia, atrial fibrillation, high grade AV block, sinus arrest with sick sinus syndrome, and terminal rhythm disorders (e.g. pulseless electrical activity, ventricular fibrillation, etc)
High grade AV block and ventricular tachycardia most likely to result in arrest in an awake patient
Atrial fibrillation, supraventricular tachycardia, and sinus arrest more likely to be associated with arrest or decompensation as a result of our interventions
What drugs can contribute to ventricular or supraventricular tachyarrhythmias?
Theophylline
Terbutaline
Phenylpropanolamine
Thyroxine supplementation
What drugs can cause or contribute to bradycardia?
Beta-blockers
Calcium channel blockers
Digoxin
When is ventricular tachycardia associated with a medium to high risk for sudden death?
When sustained (lasting >30 seconds in duration)
When is treatment of ventricular tachycardia indicated in small animals?
Indicated in an awake animals with clinical signs resulting from the arrhythmia (weakness, worsening CHF, collapse, or syncope) and in animals with sustained (>30 seconds) and rapid (>170-200 bpm) ventricular tachycardia
Treatment for Ventricular Tachycardia in Small Animals
IV lidocaine boluses followed by CRI
If ineffective then procainamide can be used
Potentially amiodarone
Chronic oral therapy
Beta blockers (sotalol, atenolol, metoprolol, propranolol) or other class I antiarrhythmic drugs, especially mexiletine where available
Treatment for Ventricular Fibrillation
Immediate electrical defibrillation
Success Rate of Electrical Defibrillation for Ventricular Fibrillation
Likelihood of success is inversely proportional to the amount of time in ventricular fibrillation
Successful defibrillation reduced by approximately 50% for every 3-5 minutes of time delay
IV amiodarone and magnesium can be used in addition to electrical defibrillation for those that are refractory to repeated attempts
Low (standard) doses of epinephrine for support of CPR are preferred to higher doses
Treatments for Supraventricular Tachycardia in Small Animals
Diltiazem
Verapamil
Propranolol
Esmolol
Caution using concurrent beta-blockers and calcium channel blockers as this can lead to hypotension or excessive bradycardia
Chronic management
Digoxin
Drug of choice when accompanied by heart failure
Calcium channel blockers (diltiazem, verapamil) or beta-blockers (propranolol, atenolol, metoprolol, sotalol)
Amiodarone
Sinus Arrest
A pause of greater than 2 P-P intervals without evidence of depolarization of the sinus node
Sick Sinus Syndrome
Characterized by bradycardia due to sinus node dysfunction, typically accompanied by short bursts of supraventricular tachycardia
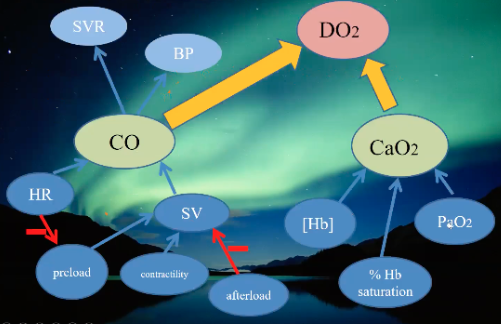
What are traditional methods of detecting altered tissue perfusion as proposed surrogates for oxygen delivery?
Physical examination, body temperature, arterial blood pressure, central venous pressure (CVP), and urine output
Have poor correlation to microcirculatory perfusion and cardiac preload and may fail to indicate early signs of global tissue hypoxia
How do you assess global oxygenation status?
Measure oxygen delivery and consumption (VO2)
Obtain these values with pulmonary arterial or central venous catheters
What are traditional means of assessing macrocirculation?
Global perfusion parameters, cardiac output, CVP, base excess, and lactate
What are noninvasive methods to detect altered microvascular circulation and occult or compensatory shock?
Near infrared spectroscopy (NIRS)
Dark field videomicroscopy
Doppler flowmetry
Gastric tonometry
Sublingual capnometry
Transcutaneous carbon dioxide measurement
Tissue Spectrometer Technology
Near infrared spectroscopy measures the absorption of infrared light (wavelengths of 700-1000 nm) by tissues to determine the oxygen hemoglobin saturation of blood in vessels less than 1 mm in diameter within the tissue
Oxyhemoglobin preferentially absorbs higher near-infrared wavelengths (800-1000 nm) whereas deoxyhemoglobin absorbs wavelengths closer to 600-800 nm
Scattering of light is one of the biggest problems encountered when utilizing NIRS technology, 80% of the light emitted is lost to scatter
Probe placement over hematomas, fat, or bone may alter tissue oxygen reading
Fluctuations in body temperature, as well as excessive movement, can lead to errors in tissue oxygen saturation (StO2) measurement
The sensor is placed over a muscle bed
The most reliable StO2 readings in people were achieved using the thenar eminence (muscle at the base of the thumb) and in dogs using the sartorius
Total Hemoglobin Index (THI)
An indicator of signal strength of near infrared spectroscopy, measures the amount of intravascular hemoglobin, intramuscular myoglobin, melanin, and mitochondrial cytochrome c oxidase
THI measurements of 5 or less indicate a weak hemoglobin signal and may lead to inaccurate StO2 readings
Vascular Occlusion Testing
Vascular occlusion testing (VOT) determines the baseline StO2 and then evaluates rate of deoxygenation and reoxygenation following occlusion of regional circulation
StO2 is continuously measured at a distal site during occlusion of blood flow using a sphygmomanometer and pressure cuff
The ischemic challenge lasts for a defined interval of time or until the StO2 meets a specific threshold
Cuff is then deflated rapidly and StO2 recovery slope obtained
May be useful when examining patients with sepsis or septic shock as they often have decreased oxygen extraction (VO2) from the tissues
Decreased oxygen consumption in patients with septic shock results in a prolonged recovery slope in the VOT
Currently recommended that a target StO2 value be used for monitoring instead of the absolute values determined by the VOT
VOT is a measure of microcirculatory reserve and not a direct representation of microcirculatory perfusion
Digital extensors are the muscle belly of choice for VOT in veterinary patients
Tissue StO2 in the Human Literature - Trauma
Low StO2 can predict the occurrence of multiple organ dysfunction after sustained traumatic shock and is more sensitive to predicting multiple organ dysfunction syndrome, the need for massive transfusions, and death in traumatized patients than other diagnostic tools
StO2 improves more rapidly than plasma lactate concentration and base deficit after adequate resuscitation which may lead to lower volumes of intravenous fluids administered to trauma patients
StO2 has been shown to be an early and accurate predictor of the need for life-saving interventions compared to plasma lactate concentration and base excess
Direct relationship between the magnitude of oxygen deficit and the risk of multiorgan failure -> treatment rationale that optimizes cardiac output and hematocrit to correct deficits in VO2 and tissue oxygen delivery
Tissue StO2 in the Human Literature - Surgery
StO2 improves with IV fluid administration prior to normalization of other traditional systemic hemodynamics (e.g. arterial blood pressure)
Lower StO2 values may be associated with increased postoperative complications such as SSI
Monitoring of StO2 levels during and after anesthetic events may be helpful to prevent postoperative complications such as SSI, duration of ICU hospitalization, and morbidity and mortality
StO2 in Human Literature - Sepsis
Due to the vast alterations in microcirculation in patients with sepsis, StO2 can be low, normal, or increased making it difficult to interpret
StO2 values have been shown to correlate to the severity of disease and to mortality in patients with severe sepsis and septic shock
Alternative monitoring sites for StO2 in septic patients due to difficult of using the thenar eminence
Knee
Masseter
Deltoid
Tissue oxygen monitoring in septic patients is controversial so can be used in conjunction with the VOT, which shows an impaired postischemic hyperemic response in patients with sepsis and septic shock
StO2 is lower in nonsurvivors than in survivors after early goal-directed resuscitation for septic shock
StO2 in Human Literature - Early Goal Directed Therapy
Early goal directed therapy (EGDT) is characterized by intensive monitoring for optimization of oxygen delivery in patients with severe sepsis or septic shock
Resuscitation endpoints include targeted values for ScvO2, arterial lactate concentration, pH, base deficit, and hematocrit
Utility of EGDT recently called into doubt
Methods of evaluating global oxygenation in EGDT resuscitation are invasive (requires central venous catheter and arterial blood pressure monitoring) so use of StO2 may be helpful
Conflicting results about utility of StO2
StO2 in Veterinary Literature
Measurements taken at the sartorius in dogs provided the most consistent readings
StO2 levels in dogs with shock were significantly different than those in normal dogs
Strong correlations are present between mean oxygen delivery index and StO2 in an experimental model of hemorrhagic shock in anesthetized Beagles
Potential Applications of StO2 in Veterinary Medicine
Definitive indications for monitoring of StO2 in clinical veterinary patients not fully described
Utility as a tool for triage and evaluation of human patients with trauma, sepsis, and surgical diseases so tissue oxygen monitoring may prove to be of benefit in parallel veterinary applications
May improve veterinary clinicians abilities to detect occult shock
Tissue oxygen monitoring may be useful to provide prognostic information and direct resuscitation in veterinary trauma patients
Comparison of StO2 and ScvO2 in septic veterinary patients would be useful to determine if StO2 can be used as a noninvasive surrogate in this population
Has not been evaluated in felines
Uses of Tissue Oxygen Monitoring
Tissue oxygen monitoring may be used to detect hypoxia in emergency settings where monitoring of initial resuscitation may aid in improving outcome
May also be helpful to identify states of hypoperfusion
Advantages of Tissue Oxygen Monitoring
Rapid
Continuous
Noninvasive
Portable
Easy to Use
Accuracy of StO2 in People
StO2 only has a moderate correlation with ScVO2 in people
Tissue oxygen monitoring should be considered in patients who may be at high risk of morbidity and mortality or where more invasive means of measuring ScvO2 or mixed venous oxygen saturation are not feasible
High StO2 levels in patients with sepsis or septic shock may be suggestive of impaired oxygen utilization
Limitations to NIRS Technology
Lack of standardized variables among machines and measurement sites
StO2 does not measure microcirculatory flow directly and therefore it is difficult to differentiate states of altered tissue oxygen consumption from states of decreased oxygen delivery
Dyshemoglobinemias can influence the StO2 level
The path length of near infrared light will vary depending on the composition or density of tissue as well as the degree of melanin so is altered in animals with different degrees of pigmentation
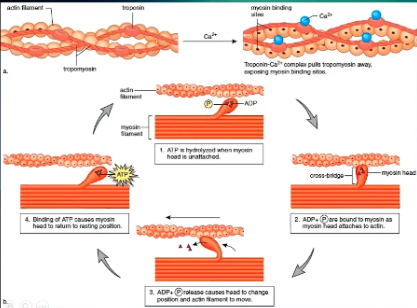
How does cardiac contraction work, actin/myosin level?
In normal resting, noncontracted state have actin and myosin tropomyosin
Tropomyosin covers the myosin binding sites
Have to move it off to allow actin and myosin to bind
Calcium binds to tropomyosin and pulls it off so myosin binding sites are open
Myosin only binds to those sites when it has ADP bound to it
When actin and myosin bind, ADP is released, causing movement of actin and myosin and get sliding of muscle filaments against each other
In order to release and get ready to do this again, ATP must bind to myosin
When ATP binds to myosin it releases actin and its ready to rebind again
ATP is dephosphorylated to become ADP/myosin complex
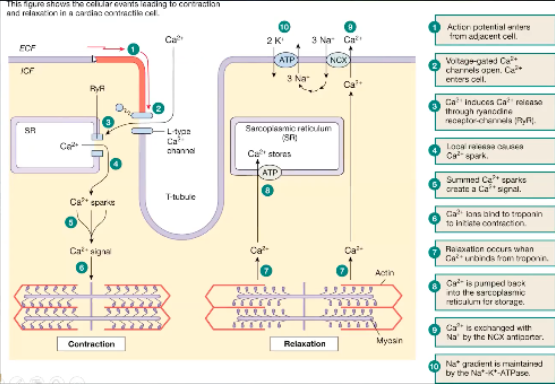
How does cardiac contraction work at the cellular level?
In order to get troponin to move off of myosin binding sites and to get any muscle contraction you need calcium inside the cell
Calcium goes into the cell through L type calcium channel
Voltage gated, won't open until there's enough calcium outside and enough positive charge to open the channel and go down the gradient
In the cell will cause release of calcium stored in the sarcoplasmic reticulum
Specifically the channel the calcium comes out is the ryanodine receptor channel
This is now called a calcium spark, when there's enough calcium spark it will bind troponin and bind off of the myosin binding site
Actin and myosin can interact with each other
When we want a relaxed state, Ca releases from troponin to allow the troponin to go cover the myosin binding sites
Ca is released from troponin and is stored back in the SR - ATP dependent channel
Some of the calcium leaves via Na/Ca transporter
NCX transporter
To have Na pumped back in to exchange Na it must be actively pumped back in via Na/K/ATPase, active process
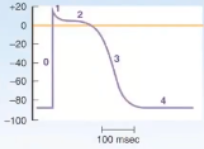
Describe the Steps of the Ventricular Action Potential
0 - sodium rushes into the cell down a gradient
2 - to keep a plateau you need + to keep going into the cell, that's Ca2+
K+ starts to go out quickly after Na+ comes in so need Ca+ to come in to keep the plateau
3 - repolarization, lots of Ca2+ out, no more Na+ in, no K+ in
4 - back to resting membrane potential
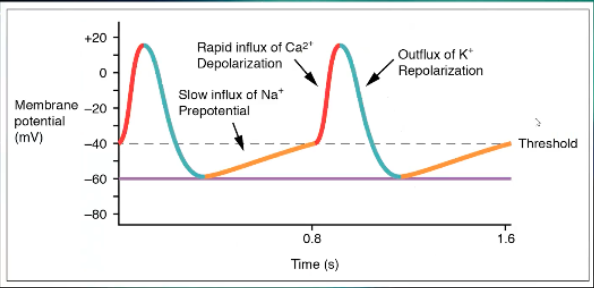
Describe the Steps of the Action Potential of the SA Node
Starts at a less negative resting membrane potential, -40, a little bit easier to get it depolarized sooner than ventricle, but does drop down to a point where it can't be depolarized again, refractory period
During refractory period, Na+ will slowly move into the cell until you get to threshold, Na+ still first step, just gradual
Sharp increase is from Ca2+ rather than causing plateau
When you repolarize its K+ leaving the cell
When you have arrhythmia when there's something affecting the SA node, A fib, SVT, most important thing to block is Ca2+, use Ca channel blockers
Draw the Wigger’s Diagram
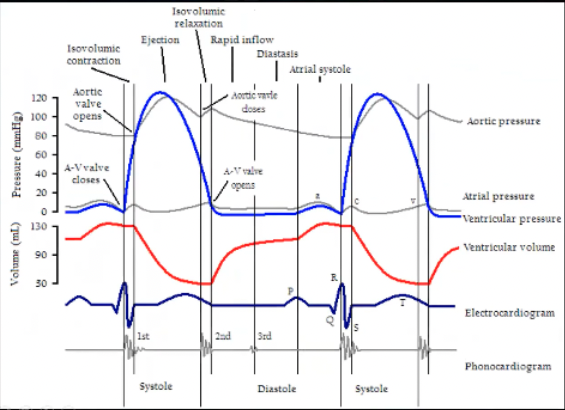
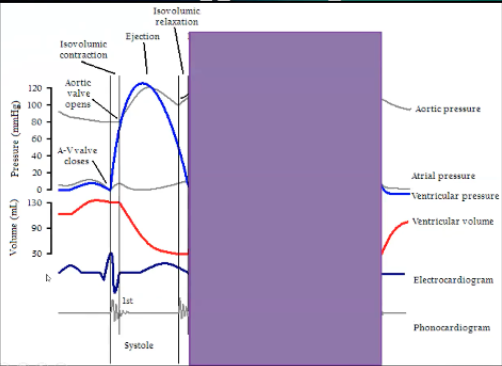
Wigger’s Digram Systole
Systole
When is systole on this diagram
Systole - peak of QRS to end of T wave
Systole starts at the top of the QRS wave at highest peak with cloving of AV valve and opening of aortic valve, moves on through blood leaving the ventricle
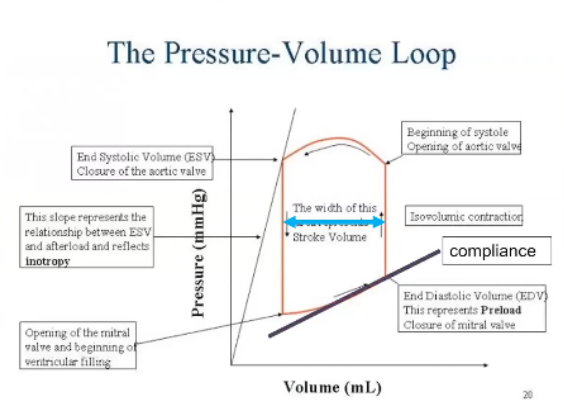
Isovolumic contraction occurs when both valves are closed, systole happens, AV valve closes cause ventricle is full, based on pressure in ventricle is equilibrated to atria or higher, isovolumic contraction squeeze the heart until pressure in ventricle exceeds that in the aorta and that opens the aorta, end of isovolumetric contraction
Then ejection phase
Ventricular pressure goes up during isovolumetric contraction until it exceeds aortic pressure, aortic valve opens
Ejection phase where it will go a little higher to get blood out, then pressure will fall a little because there's less blood, ventricular pressure falls below the aortic pressure, get closure of aortic valve, get isovolumetric relaxation where ventricle relaxes until pressure is lower than the atria, AV valves open, end of isolumetric relaxation
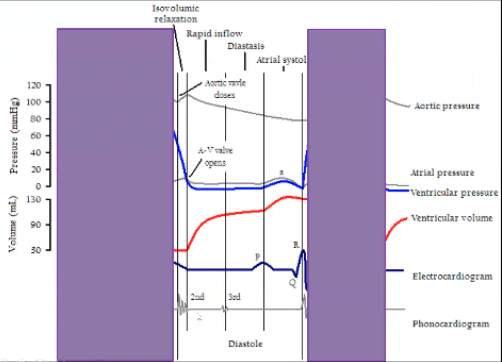
Wigger’s Diagram Diastole
Diastole
Starts during isovolumetric relaxation phase, beginning of diastole
Occurs from end of T wave until middle of the R wave
Low ventricular pressure
Ventricular volume is going up, filling ventricle
At the end of diastole get P wave and atrial contraction so atrial pressure is low and a little oomph to push blood out of the atria into the ventricle
During isovolumetric relaxation all valves are closed, at the end AV valve opens, then is open for entire diastole because allowing blood in, at the beginning of isovolumetric contraction AV valves close
Draw the Pressure Volume Loop and Label the Points
What tells the kidney to release renin?
Sympathetic stimulation
Hypotension
Decreased sodium delivery
What are the two hormones in the posterior pituitary gland?
ADH and oxytoxcin
What are the stimuli for ADH release?
Low blood pressure
High plasma osmolarity
What antagonizes ADH and the RAAS system?
ANP
Draw Vascular Smooth Muscle Contraction and Relaxation Diagram
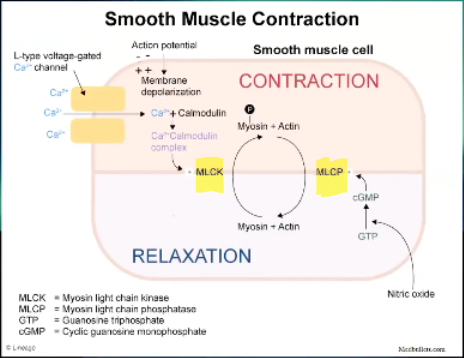
What is the process of vascular smooth muscle contraction?
Still L type voltage gated Ca channel
Ca goes into smooth muscle cell
Ca binds to calmodulin -> Ca/calmodulin complex
Stimulates myosin light chain kinase -> phosphorylates the myosin -> myosin releases the actin its bound to be able to bind again and contract
Smooth muscle contraction often happens in response to an action potential
Cell membrane depolarization will be what makes the Ca channel open
This is how epinephrine/norepinephrine and angiotensin II cause vasoconstriction
What are the major players in vascular tone?
Norepinephrine/epinephrine
Angiotensin II
Vasopressin
What is the process of vascular smooth muscle relaxation?
Relaxation happens in response to many things, NO the biggest player
NO produced by NO synthase
Diffuses into the cell, no channel
NO take GTP and turns it into cGMP -> activates myosin light chain phosphatase -> phosphatase take off the phosphorous from myosin and allows binding between myosin and actin which is released and you phosphorylate the myosin again
Causes relaxation of smooth muscle
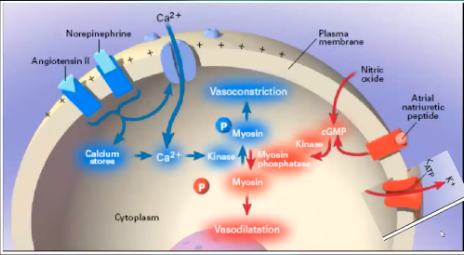
How nitric oxide and ANP cause vasodilation
Effects of Nitric Oxide on KCa Channels
Indirect effects by opening KCa channels
Direct nitrosylation of the channel
Activates cGMP-dependent protein kinase
Contributes to vasopressor resistance in septic shock
When NO binds to Kca → K+ efflux out of the cell → vasodilation
How the K+/ATP channel affects vascular smooth muscle tone
When Ca channel is activated K leaves the cell → interior of cell more negative , inside of cell hyperpolarized, makes L type Ca channel close, Ca can't get into the cell and can't cause cell to contract, can't get vasoconstrictive state, close Ca channel down and hyperpolarize the membrane
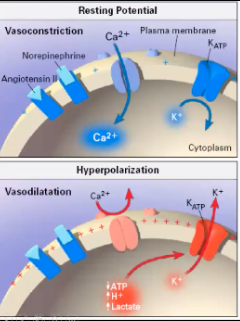
What activates the K+ ATPase in vascular smooth muscle?
Increased tissue metabolism
Hypoxia
ANP, calcitonin gene-related peptide, adenosine
Increased in septic and vasodilatory shock
Draw the “Tree of Life”
Action of a-1 Receptors
Located (central) arteries and veins → vasoconstriction
Action of a-2 Receptors
Located in the GI tract → Decreased secretions, motility, tone
Located (peripheral) arteries and veins → vasoconstriction
Actions of B-1 Receptors
Heart → increase inotropy + chronotropy
Actions of B-2 Receptors
Skeletal muscle vessels, coronary arteries → vasodilation
Actions of B-2 Receptors
Bronchial smooth muscle → relaxation
Dobutamine
Positive inotrope
Synthetic B-agonist
B1 » B2
Primarily increase cardiac contractility
Little to no change in heart rate, vascular resistance
Side Effects of Dobutamine
Arrhythmias, tachycardia, vasodilation
Can cause CNS signs in cats
MOA of Dopamine
Binds to D1 and D2 receptors as well as a1, B1, B2, a2 receptors
Low Dose Dopamine
Binds to D1 and D2 receptors
Splanchnic vasodilation, natriuresis, diuresis, variable alterations renal and GI blood flow
CONTROVERSIAL: improves urine production in oliguric renal failure
Used commonly in hypertension or pulmonary edema
What is dopamine a precursor for?
Norepinephrine
At high enough dosages, metabolized to clinically significant amounts of norepinephrine
Binds to a and B receptors → vasoconstriction
Medium Doses of Dopamine
B»a receptors
Increases cardiac contractility
Mild increase in systemic vascular resistance
High Doses of Dopamine
Primarily an a agonist
Increases systemic vascular resistance
May cause renal, GI, and cardiac ischemia
Side Effects of Dopamine
Arrhythmias, tachycardia, increased systemic vascular resistance
May decrease PaO2 → pulmonary arterial vasoconstriction
Redistribution of GI, renal blood flow → ischemic damage
Epinephrine
Higher dosages have a > B effects
Greater vasoconstrictive effects
Lower dosages do have more B effects
Greater increases in contractility with less vasoconstrictive effects
Cannot select for effects
Typically end up with both pressor and inotropic effects with epinephrine
Side Effects of Epinephrine
Increased oxygen consumption by tissues
Severe vasoconstriction (decrease blood flow to tissues)
Ischemia more common in the GI tract, kidneys, liver
Norepinephrine
Primarily a > B effects
Mainly increase systemic vascular resistance (small increases in HR only)
May increase blood flow to the heart, kidneys without ischemia in other locations
Hope is to vasoconstrict and push blood to some of the tissues
Where is vasopressin synthesized and stored?
Synthesized by magnocellular neurons in the hypothalamus
Stored in the posterior lobe of the pituitary gland