Lecture 18: Failures of Host Defense Mechanisms
1/68
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
69 Terms
Types of immunodeficiencies
Primary immunodeficiencies
secondary immunodeficiencies
combined immunodeficiencies
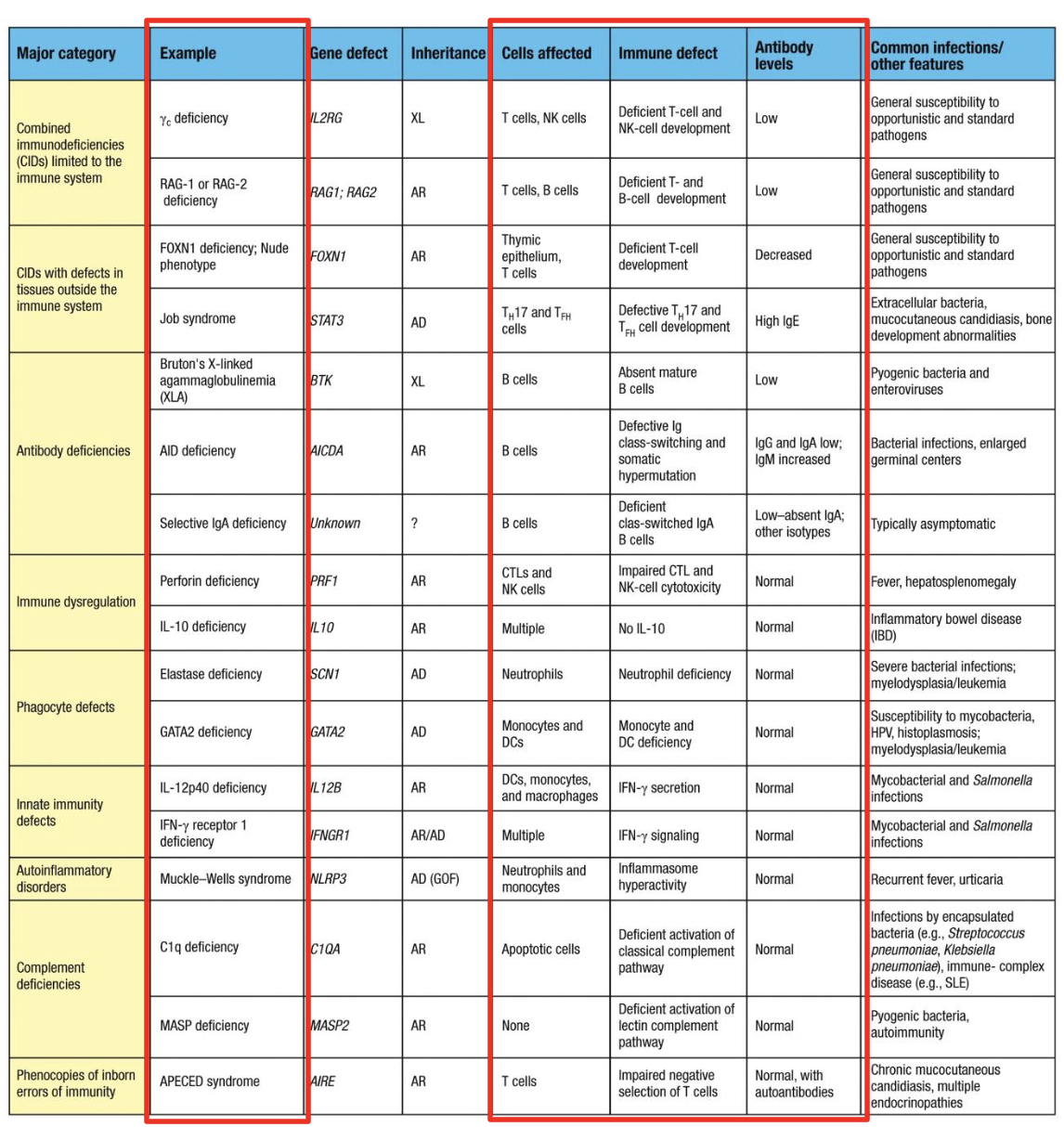
Primary Immunodeficiencies
PIDS (or congenital) are a heterogenous group of inherited disorders with defects (genetic mutations) in one or more components of the immune system
PIDs are not as rare as once believed
Infections are hallmarks
Secondary Immunodeficiencies
Non-inherited, acquired
caused by environmental factors such as malnutrition, age, infection, irradiation, chemotherapy, or exposure to toxins
infections are hallmarks
combined immunodeficiencies
impairments in both B-cell and T-cell function due to inherited mutations
what are primary immunodeficiencies caused by
caused by inherited gene variants that typically cause recurrent infections in early life
pyogenic, or pus-forming, bacteria →
a defect in antibody, complement, or phagocyte function
persistent fungal or viral infection→
defect in T-cell function
how many people are affected by primary immunodeficiencies
1 in 1000 people in the US - 80% of people are <20 yo
how are primary immunodeficiencies often inherited
often inherited in X-linked recessive manner - 70% cases among males
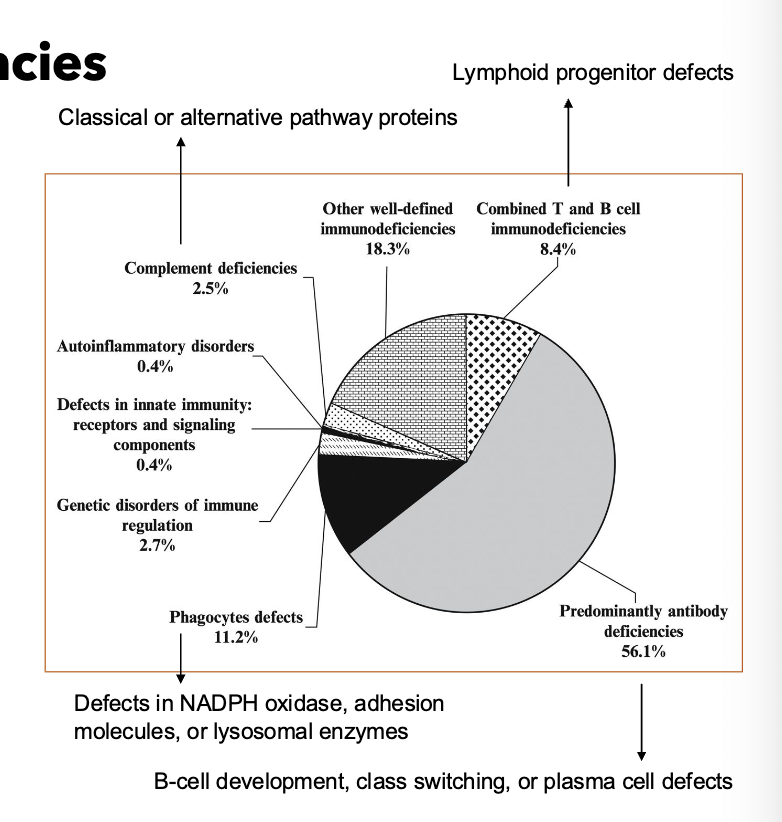
human immunodeficiency syndromes how many categories
9 categories
decreased T cells →
viral/fungal susceptibility
decreased B cells →
pyogenic bacterial infections
decreased phagocytes
abscess formation, catalase + infections
decreased complement
Neisseria infections
SCIDs linked to what
T-cell and B-cell Development
what is SCID
the most severe form of primary immunodeficiency
what is SCID characterized by
defective T-cell development, with secondary impairment of B and NK cells
how does SCID present
in the first months of life: failure to thrive, chronic diarrhea, oral thrush, persistent viral or fungal infections
is SCID fatal
fatal if untreated; curable by hematopoietic stem cell transplantation (HSCT) or gene therapy
Autosomal recessive SCID
Adenosine deaminase (ADA) deficiency → disrupts S-phase of cell cycle → improper lymphocyte development → lack of circulating T and B cells
causes SCID in infancy - must be treated with bone marrow transplantation
Omenn Syndrome
partial loss of V(D)J recombinase activity through mutations in at least one of the RAG1 or RAG2 alleles
peripheral T cells are autoreactive
Autosomal recessive SCID - both boys and girls are affected
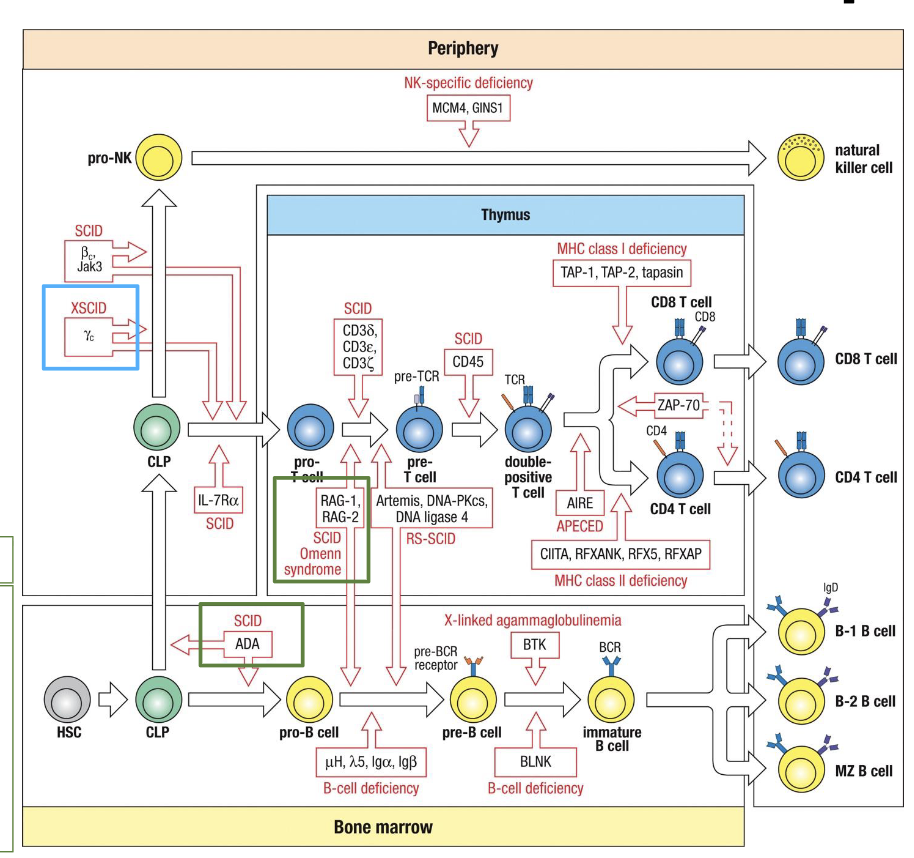
X-linked SCID
X-chromosome recessive inherited immunodeficiency that presents with virtual lack of peripheral blood T cells and NK cells. B cells are usually present but Ig production is reduced
what is X-linked SCID caused by
caused by mutation of the IL-2RG, the gene encoding the IL common gamma chain (gc) of the cytokine receptors for IL-2, -4, -7, -9, -15, and -21
what percent of cases of SCID does X-linked SCID account for
incidence of ~1:100,000 male births - accounts for approx half the cases of SCID
in normal cytokine signaling, yc pairs with what
specific ab receptor chains → JAK1/JAK3 → STAT activation → lymphocyte development and proliferation
in IL2RG mutated cells, what happens to these pathways
these pathways are nonfunctional:
IL-7/IL-7Ryc → required for T-cell differentiation in thymus
IL-15/IL-15Ryc → required for NK-cell differentiation
immunodeficiencies that alter T-cell development and function
FOXN1 mutation
DiGeorge syndrome
Bare Lymphocyte syndrome
FOXN1 mutation
lack of thymic function - abnormal T cell development
DiGeorge syndrome
22q11.2 deletion
small portion of ch22 deleted
thymus is absent - abnormal T-cell development and function
very difficult to treat
Bare Lymphocyte Syndrome (BLS)
MHC Class II deficiency or MHC Class I deficiency
mutations in TAP genes - improper activation of CD8 T cells (MHC class I deficiency)
mutation in transcription factors responsible for MHC class II expression - improper activation of CD4 T cells (MHC Class II deficiency)
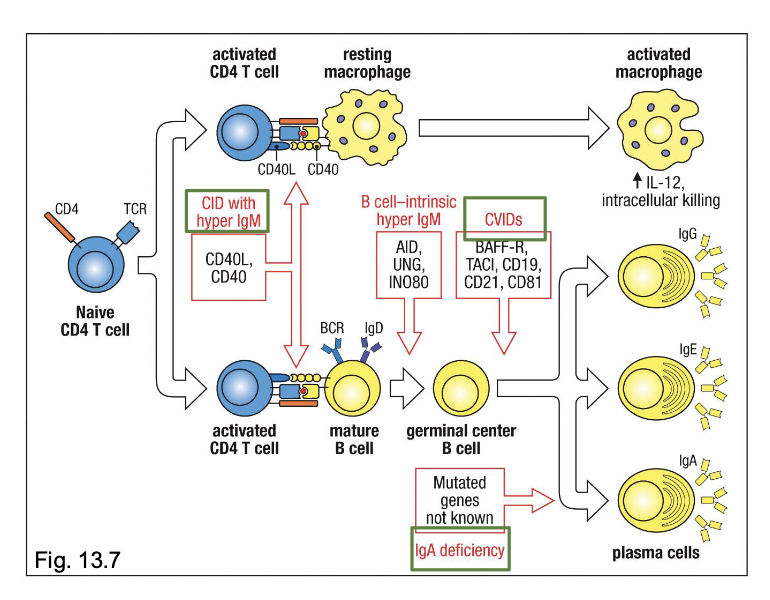
Immunodeficiencies that alter B-cell development
X-linked Agammaglobulinemia (XLA)
Hyper IgM syndrome
Common Variable (CVID)
X-linked Agammaglobulinemia (XLA)
failure of B-cell precursors to mature into B cells and then plasma cells
Mutation in BTK gene located on C chromosome
BTK transmits signal from pre-BCR necessary for maturation beyond pre-B stage
without BTK signalling, B-cell development arrests in the bone marrow → no mature B cells or plasma cells
low levels of all isotypes
reduced B cell numbers
Hyper IgM syndrome
caused by mutation in the CD40 ligand gene
CD40L expressed on activated CD4+ T cells
Affects Igs and impacts only males
low levels of serum Igs
IgG, IgA, IgE are low
IgM may be low, normal, or elevated
heightened susceptibility to infections that require high-affinity, class-switched antibodies for clearance
Common Variable (CVID)
most common form of PID
variable deficiencies in 2 or more Ig isotypes
low IgG and IgA, with or without low IgM
defective B-cell activation and plasma-cell differentiation despote normal numbers of mature B cells
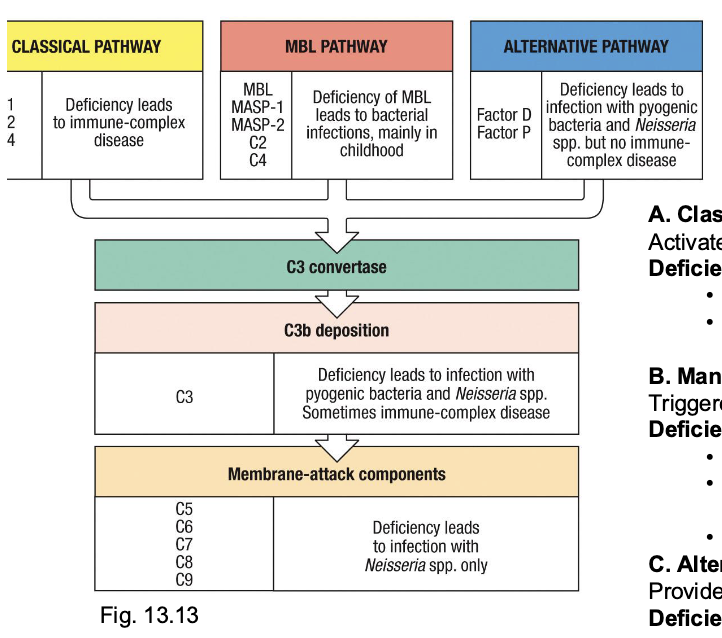
Defects in the complement system
account for ~2-5% of PIDs
a. Classical pathway (C1q, C1r/s, C2, C4)
activates via ag-antibody complexes
deficiency phenotype:
failure to clear immune complexes → lupus-like autoimmunity
recurrent sinopulmonary infections (streptococcus, Haemophilus)
B. mannose-binding lectin pathway
triggered by lectin recognition of microbial carbohydrates
Deficiency (MBL, MASP-1, MASP-2)
common in children (~5-10% population heterozygous)
usually mild → recurrent bacterial infections in infancy/childhood, esp respiratory
often subclinical in adults
c. Alternative pathway (Factor D, Factor P, Properdin)
provides constant surveillance (“tick-over”
Deficiency:
reduced opsonization of encapsulated bacteria
predisposes Neisseria and pyogenic infections, but not to immune complex disease (since no immune complex clearance role)
Immunodeficiencies of innate immune system
Chronic Granulomatous Disorder (CGD)
Leukocyte Adhesion Deficiency (LAD) I
Chediak-Higashi Syndrome
Chronic Granulomatous Disorder (CGD)
caused by defects in phagocytes
phagocytic cells cannot kill certain pathogens → form granulomas
defective production of ROS needed for killin
patients vulnerable to severe recurrent bacterial and fungal infections
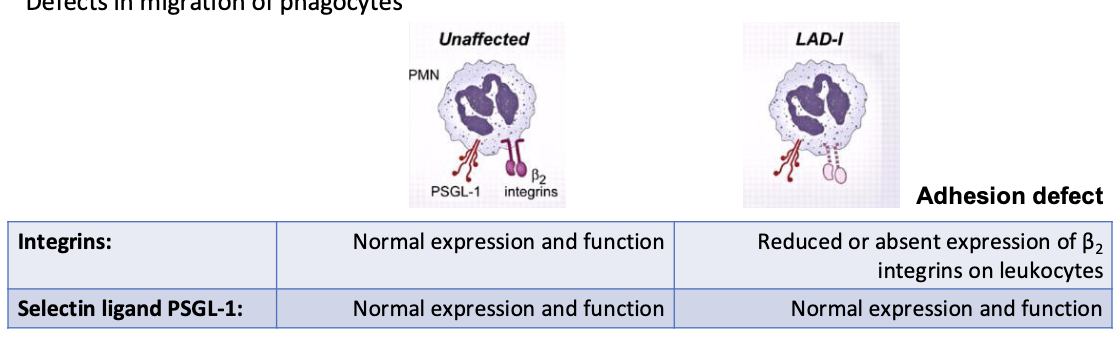
Leukocyte Adhesion Deficiency (LAD) I
defects in migration of phagocytes
reduced or absent expression of B2 integrins on leukocytes
patients deficient in expression of the 3 integrins containing CD18 (LFA-1 (CD11a/CD18) MAC-1 (CD11b/CD18), Gp 150/95 (CD11c/CD18)
Chediak-Higashi Syndrome
defects in phagocytes
impaired lysis of phagocytosed bacteria → recurrent bacterial respiratory and other infections and oculocutaneous albinism
mutation in CHS gene → affects synthesis of storage/secretory granules in various immune cells
abnormal NK cell function
defective lysosomal function in macrophages, DCs and neutrophils
hypopigmentation of skin, eye, and hair
Evasion of the Host Immune System
Mechanism of Immune Evasion - Genetic Variation
Antigenic drift and antigenic shift
antigenic variation
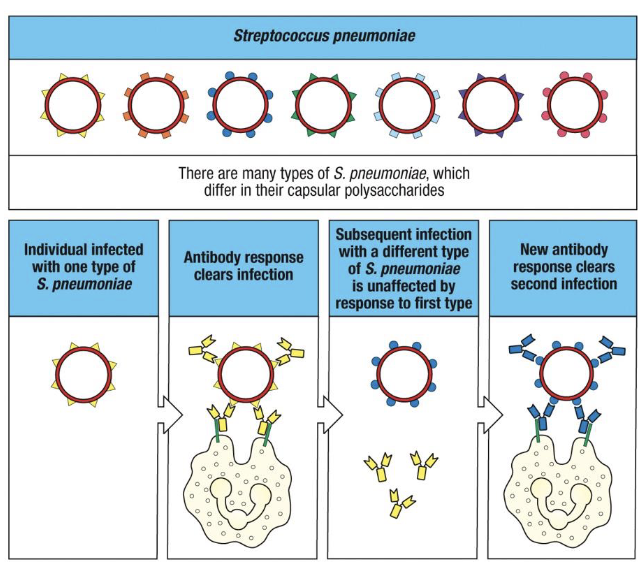
pathogen express different surface antigens without changing the bacterial genus and species
indeed, many species have multiple, serotypes, which differ on their surface and thus are not recognized by the same immunoglobulins
vaccines target multiple capsule types to broaden coverage
antigenic drift
Accumulation of point mutations during viral RNA replication (error-prone RNA polymerase)
gradual changes in hemagglutinin (HA) or neuraminidase (NA) epitopes
occurs continually → leads to seasonal influenza epidemics
pre-existing antibodies become partially ineffective

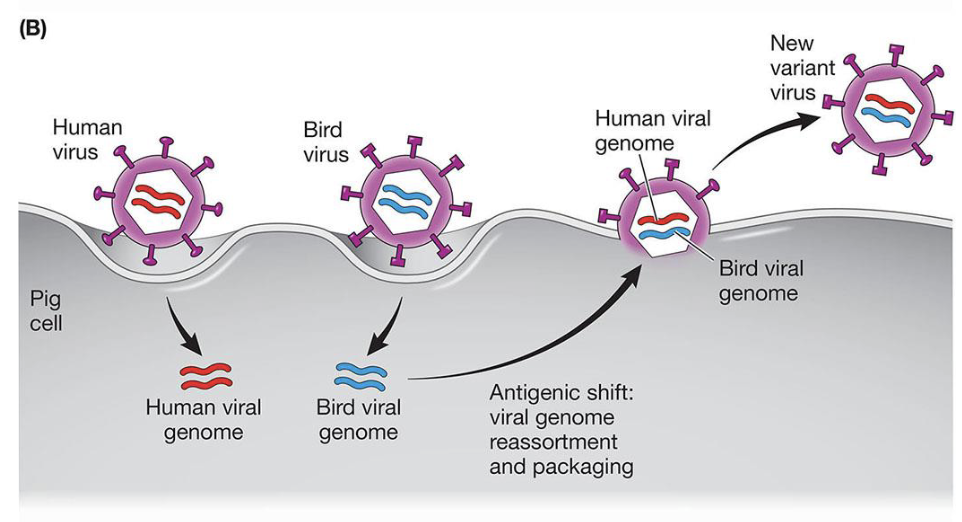
Antigenic Shift
Reassortment of viral genome segments between human and animal influenza strains in a co-infected host (e.g. pig, bird)
produces novel HA or NA combinations that are entirely new to human immunity
responsible for pandemics (H1N1, H2N2)
Viral Latency
state of nonproductive infection where the viral genome persists in host cells but no new virions are produced → allows lifelong persistence without continuous immune activation
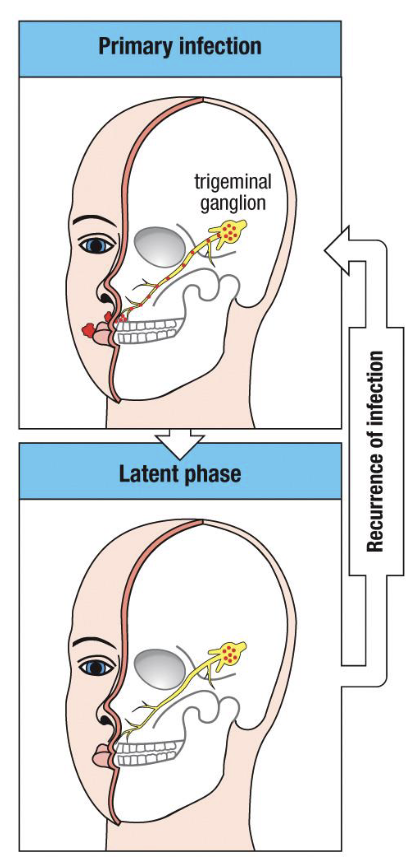
The HSV step 1: primary infection
HSV-1 (oral) or HSV-2 (genital) infects epithelial cells at the mucosal surface
viral replication produces vesicular lesions and triggers a strong adaptive immune response
virus then enters sensory nerve endings and travels retrograde to the trigeminal (or sacral) ganglion
Step 2: latent phase
viral genome maintained as episomal DNA within neuronal nuclei
no viral protein expression except LATs, which suppress apoptosis and maintain latency
no cytolysis → neurons survive → immune system cannot detect infected cells
(this is immune evasion by silence and sequestration)
Step 3: reactivation
triggered by stress, UV exposure, fever, or immunosuppression
virus travels anterograde down the same axon to the peripheral site
local epithelial replication → recurrent cold sore or genital lesion
typically milder and shorter due to memory T- and B-cell response
Acquired Immune Deficiency Syndrome
The incidence of new HIV infection is increasing more slowly in many regions of the world, but AIDS is still a major disease burden
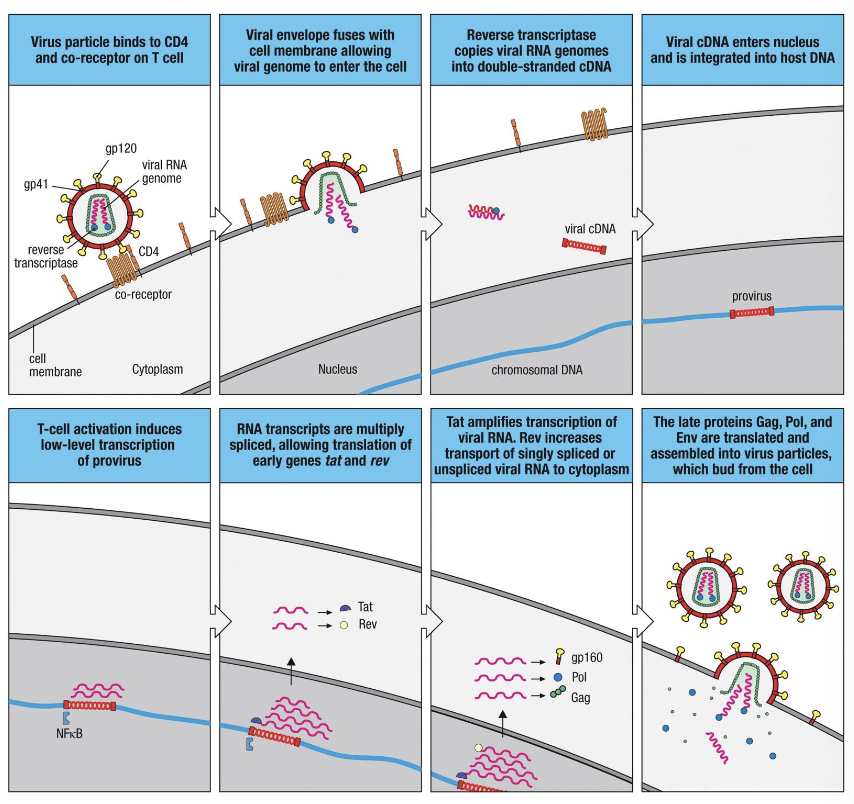
The virion of HIV
HIV is an enveloped, positive sense, ssRNA retrovirus belonging to the Lentivirus genus
what does HIV infect
infects CD4+ T cells, macrophages, and DCs
how does HIV cause AIDs
by progressive depletion of CD4+ T cells and immune dysfunction
what is the viral envelope embedded with
viral glycoprotein spikes-gp120 and gp41, which form the env complex
what does gp120 bind to
gp120 binds to CD4 receptor and co-receptors CCR5 or CXCR4 on target cells → mediates attachment
gp120 function in life cycle
recognition of CD4, CCR5 and CXCR4 receptors
what does the HIV life cycle show
the HIV life cycle shows how the virus replicates within one cell, while the infection course shows what happens when they cycle repeats billions of times across the body, leading to immune exhaustion and AIDS if left untreated
The life cycle of HIV
Typical Course of Untreated HIV Infection
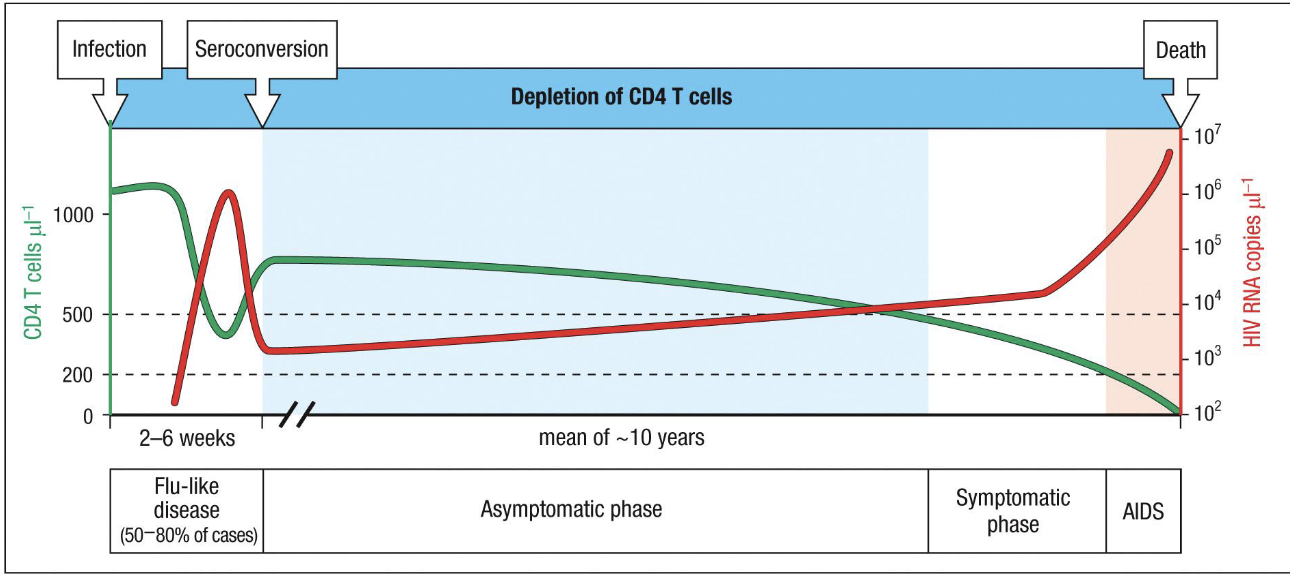
acute primary infection (2-6 weeks post exposure)
viral load spikes dramatically (up to 107 copies/microL)
CD4+ T cells plummet due to direct infection and cytolysis in mucosal tissues
symptoms: flu-like illness in ~50-80% of patients (fever, rash, pymphadenopathy)
Host immune system responds: cytotoxic CD8+ T cells kill infected cells, neutralizing antibodies appear (seroconversion)
outcome: partial control of viremia → decline in viral load → recovery of CD4 count to a lower steady state level
Clinical Latency/asymptomatic phase (~8-10 years)
Virus persists at low steady-state levels due to:
continuous replication in lymphoid tissues
formation of long-lived latent resevoirs in memory CD4+ T cells and macrophages
CD4+ T cells slowly decline due to chronic activation and turnover
patients remain clinically well, but immune system is gradually eroding
viral load remains stable because replication = clearance
symptomatic phase → AIDS
When CD4+ T-cell count <200/microL, host immunity fails
viral load surges, indicating immune exhaustion
opportunistic infections and cancers appear
death occurs from infection or malignancy unless treated
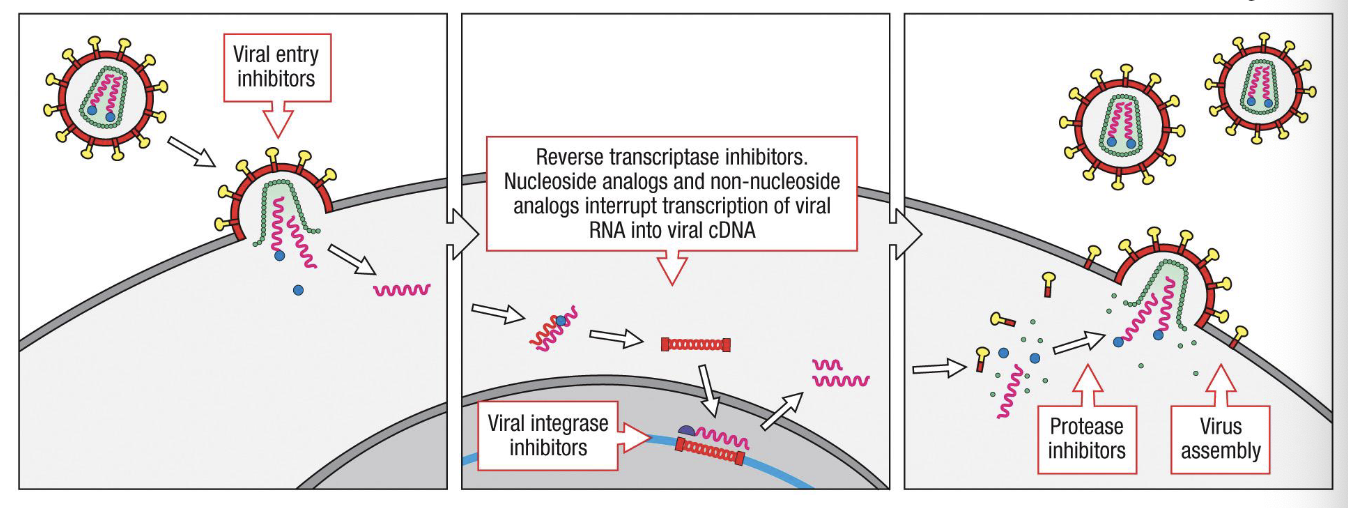
Targets for interfence with HIV life cycle
In principle, HIV could be attacked by therapeutic drugs at multiple points in its life cycle: virus entry, reverse transcription of viral RNA, insertion of viral cDNA in to cellular DNA by the viral integrase, cleavage of viral polyproteins by the viral protease, and assembly and budding of infectious virions
As yet, only drugs that inhibit reverse transcriptase and protease activation have been developed. Combination therpay using different kinds of drugs is more effective than using a single drug