M2L8: Cell death pathways
1/26
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
27 Terms
What are the morphological changes involved in apoptosis?
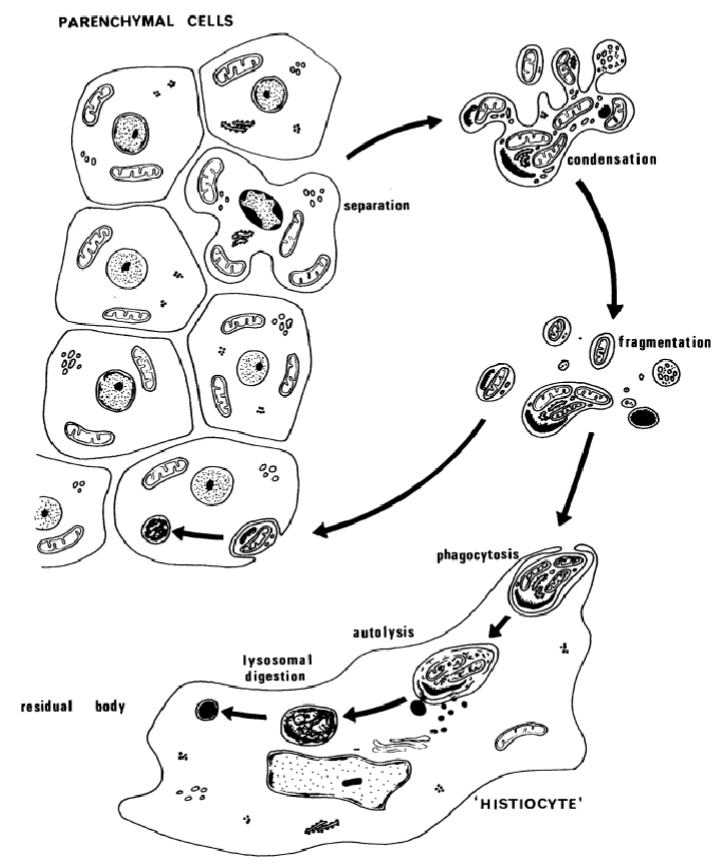
Separation from other cells
Membrane blebbing
Cell shrinkage
Nuclear/chromatin condensation
DNA fragmentation
Compare apoptosis vs necrosis
Necrosis: passive, cell swelling, membrane rupture, inflammation
Apoptosis: active, cell shrinkage, intact memnbrane, no inflammation, nuclear condensation, caspase-dependent
What are the steps in the extrinsic pathway of apoptosis?
Extrinsic apoptosis is initiated by the extracellular microenvironment and driven by death receptors
Surface receptor interaction with death ligands leads to the assembly of the death-inducing signalling complex (DISC) which is a dynamic multiprotein complex at the intracellular tail of the death receptor (death domain)
Death receptors contain a cytoplasmic domain called the death domain (DD) which helps transmit death signals from the cell surface to intracellular signalling pathways
During FasL and FAS binding, Fas-associated proteins with death domains (FADD) are recruited and during TNF and TNFR binding, a TNFR1-associated death domain protein (TRADD) is recruited with FADD
Adaptor proteins exhibit appropriate death domains to bind to their corresponding receptor and they then recruit procaspase-8 by dimerising the death effector domain (DED)
Results in DISC formation and caspase-8 is thus cleaved and activated
Active caspase-8 initiates apoptosis by cleaving and activating executioner caspase-3
What are the steps in the intrinsic pathway of apoptosis?
Intrinsic apoptosis (mitochondrial pathway) is initiated by microenvironment triggers like DNA damage or withdrawal of growth factors
Apoptotic cells retain their plasma membrane and metabolic activity during the apoptotic process due to the clearance by macrophages and other phagocytic cells via efferocytosis
At the end stage, apoptotic cells have a complete breakdown of the plasma membrane and acquire a necrotic morphotype (secondary necrosis)
Intrinsic apoptosis is irreversible and regulated by pro- and anti-apoptotic members of the Bcl2 family of apoptosis regulator proteins
The first step is widespread mitochondrial outer membrane permeabilisation (MOMP) which is mediated by Bcl2-associated X apoptosis regulator (BAX) and/or Bcl2 antagonist/killer (BAK)
BAX continuously travels between the outer mitochondrial membrane and the cytosol in an inactive form whereas BAK is located in the outer mitochondrial membrane
When apoptosis is triggered, BAX ceases to retranslocate, and BAX and BAK are directly or indirectly activated by pro-apoptotic proteins such as BID and BAD
The apoptogenic factor are released, including cytochrome C and second mitochondrial activator of caspases (SMAC)
Cytochrome C binds to apoptotic peptidase activating factor 1 (APAF1) and pro-caspse 9 to form the supramolecular complex apoptosome
The apoptosome activates caspase 9, which then catalyses the proteolytic activation of the executioner caspase 3
SMAC associates with the inhibitor of apoptosis (IAP) protein family to regulate apoptosis
Activation of executioner caspases triggers morphological and biochemical changes in the cell, including DNA fragmentation, phosphatidylserine (PS) exposure and formation of apoptotic bodies
Caspase 3 catalyses the proteiolytic inactivation of DNA fragmentation factor subunit alpha (aka. inhibitor of CAD, ICAD) and releases the catalytic activity of caspase-activated DNAse (CAD) to trigger DNA fragmentation
What is included in the Bcl-2 family?
Anti-apoptotic proteins (eg. Bcl-2, BcL-xl)
Pro-apoptotic proteins (eg. Bax, Bak)
BH3-only proteins (eg. Bid, Bad)
Compare initiator and effector capsases
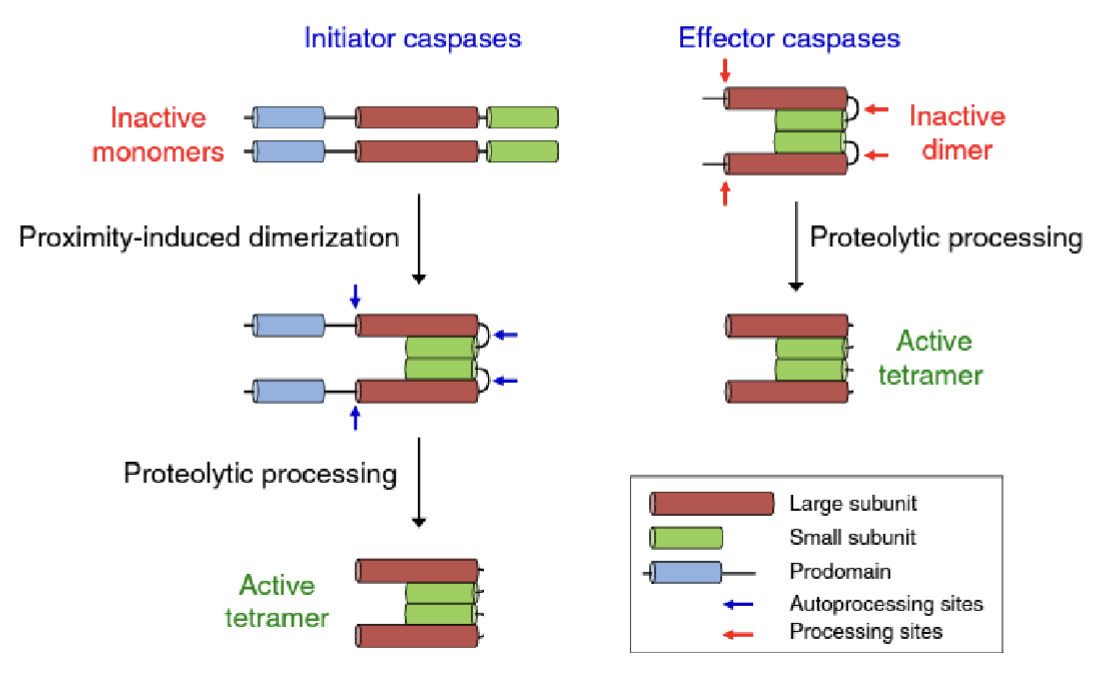
Initiators - caspase 2, 8, 9, 10
Exist as inactive monomers which undergo proximity induced dimerisation
Dimers undergo autocatalytic proteolytic processing to form the active tetramer
Executioners (effectors) - caspase 3, 6, 7
Exist as inactive dimers which get cleaved by initiator caspases to form the active tetramer
Caspase 8 activation can link extrinsic and intrinsic pathways
What is the role of p53 in the intrinsic apoptotic pathway?
p53 activates BIM, PUMA, and NOXA (BH3 only proteins within Bcl-2 family) which inhibit pro-survival Bcl-2 proteins including Bcl-XL, MCL-1 and Bcl-2
This disinhibits apoptosis effectors Bax and Bak to activate MOMP and apoptosome formation
What is the role of p53 in modifying the apoptotic response?
Activates other genes which fine-tune apoptosis, such as…
FAS/DR5 - death receptors (extrinsic pathway)
ZMAT3 - unclear role, modifies apoptotic signalling
miR-34a - inhibits translation of anti-apoptotic Bcl-2
BTG2/PLK2 - stress response genes
What is Venetoclax (venxlexta)?
Bcl-2 inhibitor for chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL)
Can be in combination with azacytidine/decitabine/low-dose cytarabine for AML
What is the function of autophagy?
Provides nutrients to maintain cell function by degrading large molecules, organelles and proteins
Removes damaged organelles, misfolded proteins, lipid droplets —> cell homeostasis
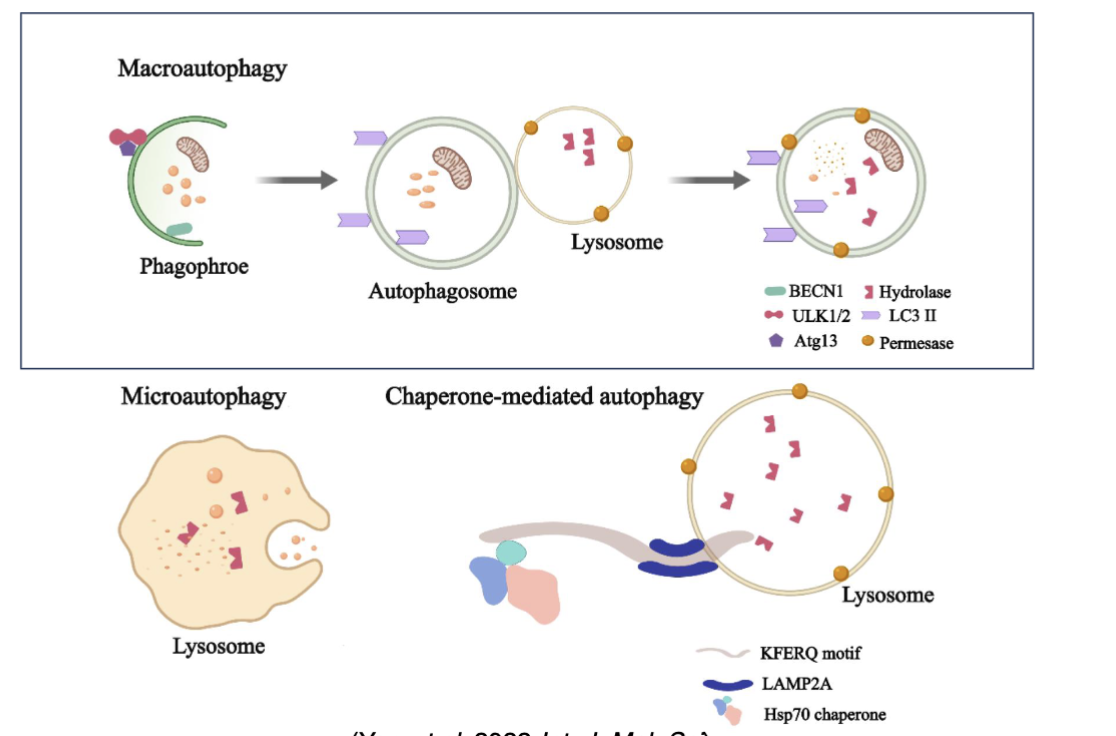
What are the types of autophagy?
Macroautophagy (most common) - characterised by formation of autophagosomes
Microautophagy - endosomes or lysosomes directly engulf and degrade autophagic cargo
Chaperone-mediated - cytosolic proteins are delivered to lysosomes for degradation by a process involving chaperones which bind proteins via specific targeting motifs
What are the steps of macroautophagy?
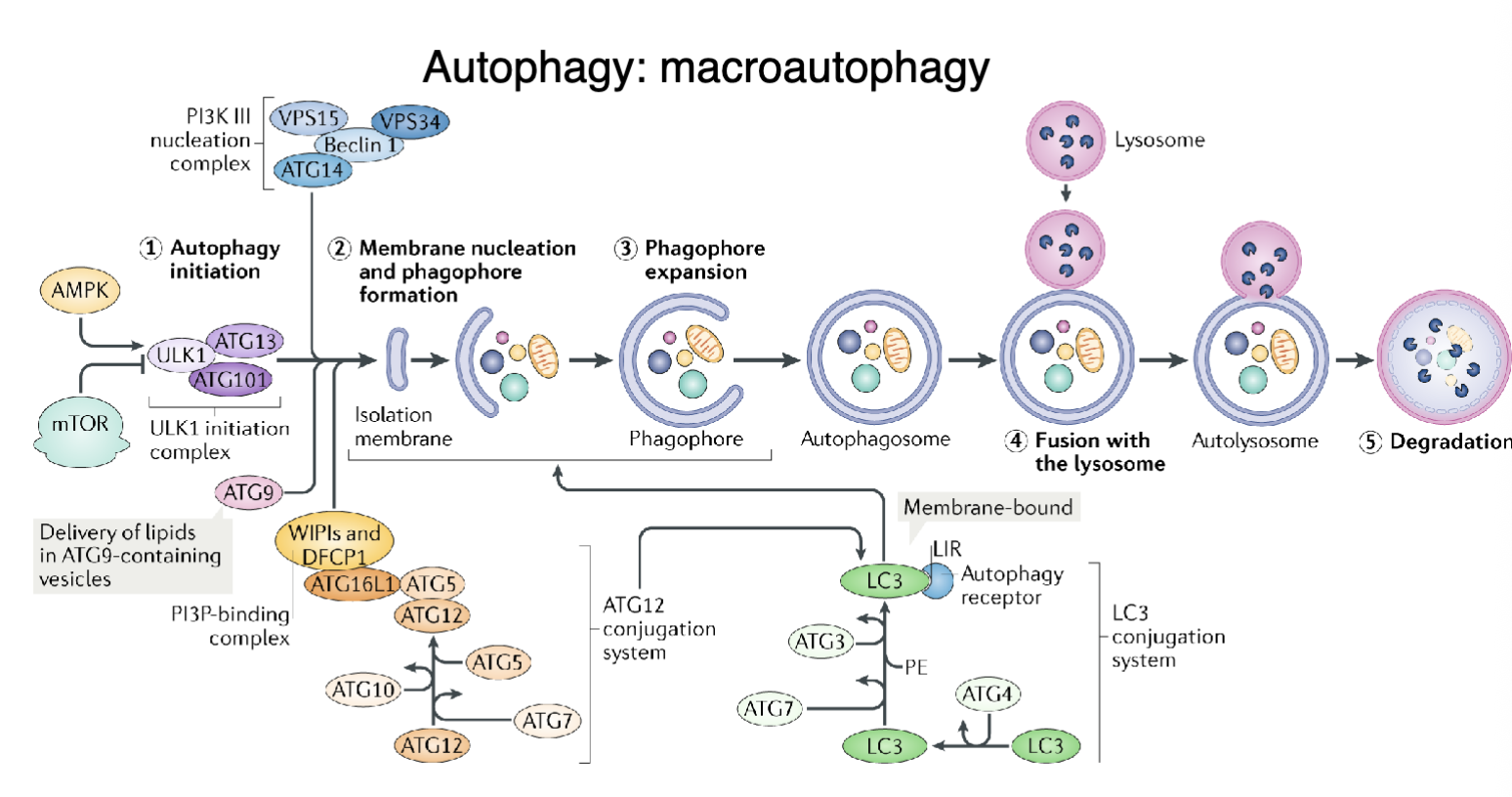
Induction and nucleation - energy/nutrient stress activates AMPK/inhibits mTOR to form the ULK1 initiation complex and the formation of a membrane ‘seed’ (isolation membrane) at the ER or other organelles
Elongation - isolation membrane expands around cytoplasmic material and incorporates LC3 (cleaved from LC3-I to LC3-II) to capture cargo
Closure and maturation - membrane edges fuse to form autophagosome
Fusion: mature autophagosome fuses with a lysosome to form and autolysosome
Degradation and recycling - lysosomal enzymes break down cargo and the resulting metabolites are recycled into the cytoplasm for energy and biosynthesis
What is the role of autophagy in cancer?
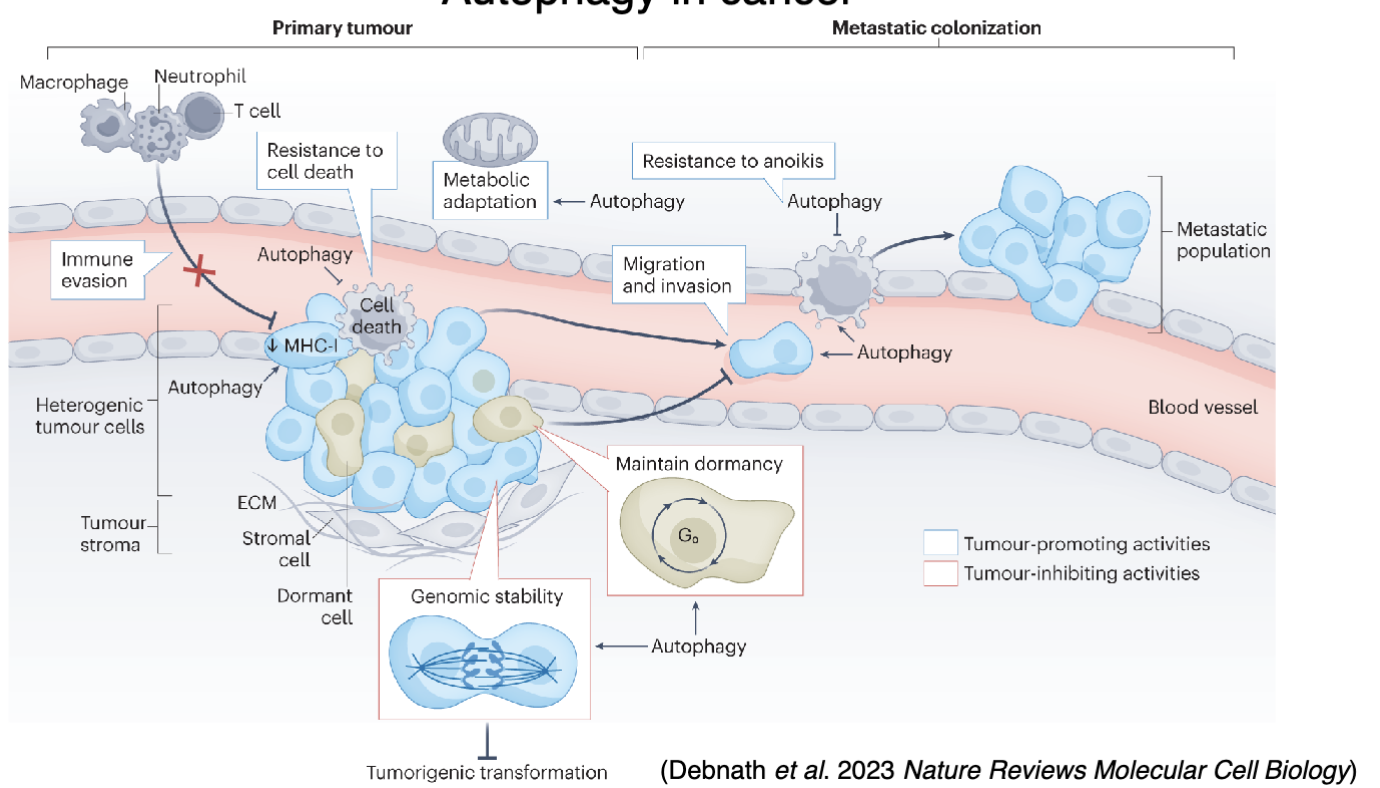
Can be pro- or anti-cancer
Autophagy can sense genomic instability and help maintain genomic integrity
Autophagy may also help maintain dormancy
Autophagic MHC-I degradation for immune escape
Autophagic degradation of damaged organelles to provide nutrients and energy for the stress response
Can promote migration/invasion
Can promote resistance to anoikis to prevent cell death when metastasising cells detach from the primary tumour and migrate to distant sites
Can maintain dormancy, which may be harmful especially at distant metastatic sites
How can autophagy be targeted in treatments?
Bafilomycin is a well known autophagy inhibitor
In clinic chloroquine and hydroxychloroquine (anti-malaria and anti-inflammatyory drugs) can inhibit fusion of autophagosomes with lysosomes and theri degradation
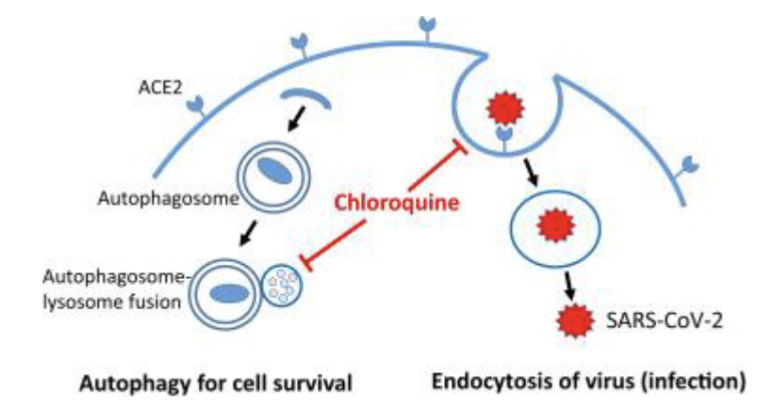
When combining chloroquine + radiation, overall survival does not notably change but helps brain metastases progression free survival (cure vs quality of life)
What are the steps in ferroptosis?
A cell death pathway which requires iron, lipid peroxidation and defective/insufficient antioxidant response
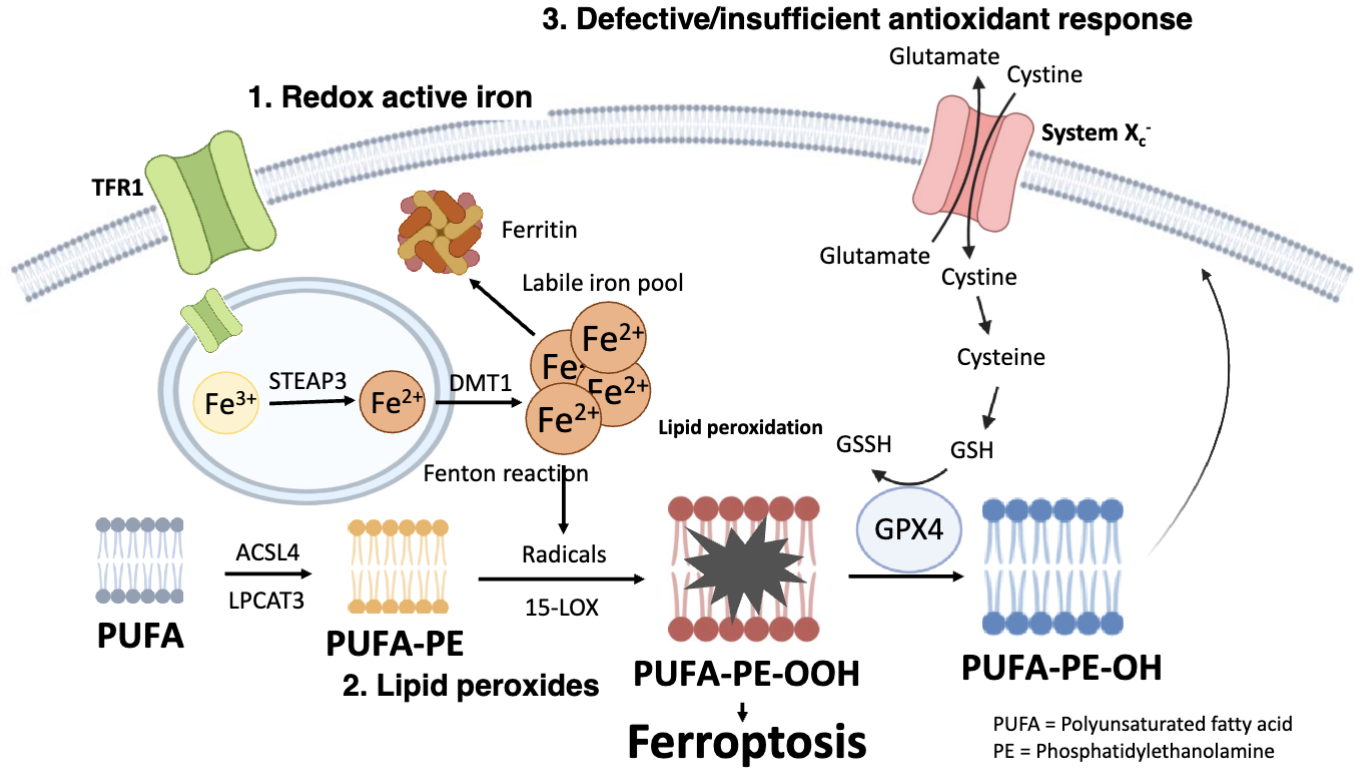
Cells take up iron via TRF1 receptor which imports Fe3_ bound to transferrin
In endosomes, STEAP3 reduces Fe3+ to Fe2+
DMT1 moved Fe2+ into the cytosol, producing the labile iron pool
Some iron is stored as ferritin and excess Fe2+ participates in Fenton reactions which generates reactive hydroxyl radicals, initiating lipid peroxidation
Polyunsaturated fatty acids (PUFAs) are susceptible to oxidation and enzymes such as ACSL4 and LPCAT3 incorporporate PUFAs into phosphatidylethanolamine (PE) in the membrane to form PUFA-PE
PUFA-PE is oxidised by 15-LOX and hydroxyl radicals to form lipid hyperoxides (PUFA-PE-OOH)
PUFA-PE-OOH accumulation causes membrane damage and ferroptosis
When System Xc- (antioxidant response) is inhibited or GPX4/GSH is low, lipid peroxides accumulate, causing ferroptosis
What happens in system Xc-?
The system Xc- antiporter imports cystine in exchange for glutamate
Cystine is converted to cysteine and used to synthesise glutathione
GPX4 uses GSH to reduce PUFA-PE-PE-OOH to PUFA-PE-OH, preventing lipid damage
How may ferroptosis be specific for targeting aggressive and invasive tumour types?
ZEB1 is an important transition factor for EMT induction which also upregulates PUFAs
GPX4 inactivation causes PUFA accumulation and lipid peroxidation leading to drug resistance
Drug resistant tumour cells often also have higher PUFA content and depend on GPX4 to manage redox stress
Inducers of ferroptosis which inhibit GPX4 can therefore cause ferroptosis in drug resistant cells
What are the morphological features of ferroptosis?
Mitochondrial condensation/shrinkage, membrane rupture
ferroptosis chain reaction
Death of one cell can be propagated to others
Explain the role of autophagy in ferroptosis
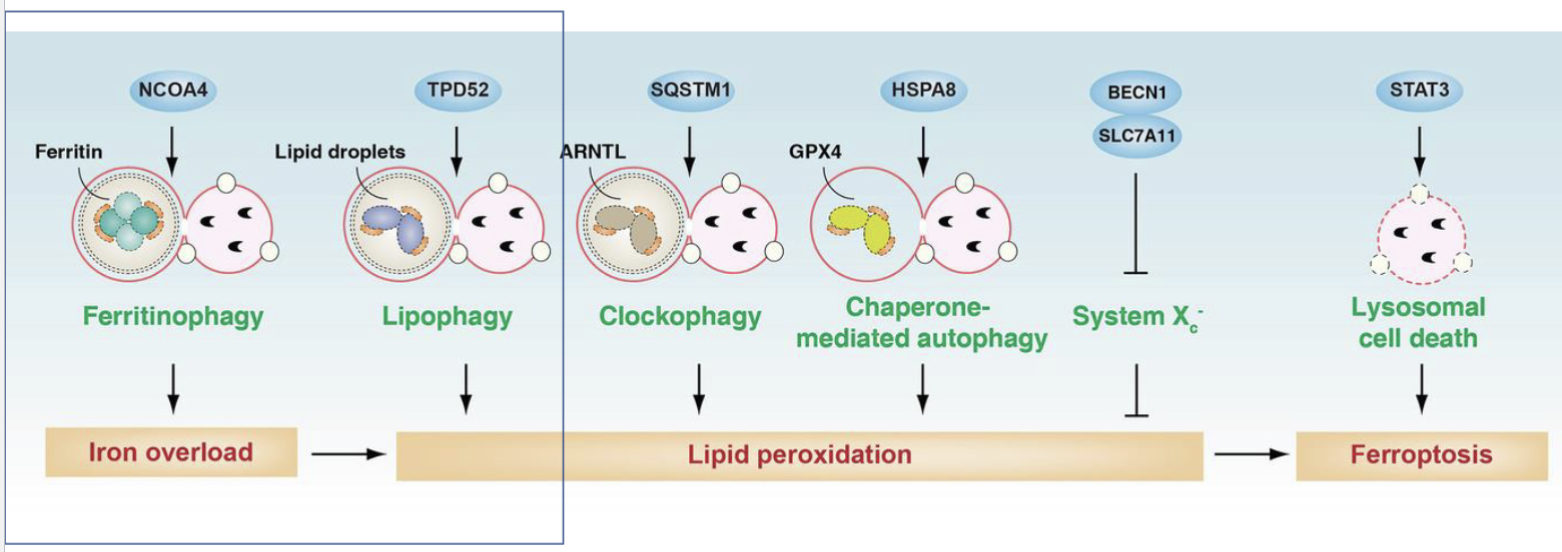
Autophagy mediated by NCOA4 can degrade ferritin and release iron into the cytoplasm to trigger ferroptosis
Lipid can be stored as lipid droplets within cells which can be broken down by autophagy nediated by TPD52, releasing free fatty acids including PUFAs which are substrates for lipid peroxidation
ARNTL is a part of the circadian clock which regulates antioxidant defence that can be degradaded by SQSTM1 mediated autophagy
HSPA8 in chaperone mediated autophagy can regrade GPX4
What are some markers that can be used to measure ferroptosis?
Ferroptosis lacks specific markers to measure it
Lipid peroxidation is the main indicator
Cell death is another indicator
When using ferroptosis inducers (molecule being tested for whether it causes ferroptosis) and inhibitors simultaneously, the amount of rescue can indicate how much ferroptosis was happening
4-HNE is a widely used lipid peroxidation marker in tissue staining
What is the role of MAFF in ferroptosis?
MAFF is an important factor regulating ferroptosis via lipophagy reaction
Given that there are no approved inducers of ferroptosis in clinic, how can existing drugs be repurposed for this?
Sulfasalazine, sorafenib, and olaparib inhibit system Xc-, reducing cystine uptake → lowering GSH → weakening GPX4 activity
Artesunate depletes reduced GSH or inhibits GPX4 activity directly
Ionising radiation causes lipid peroxidation or inhibits the antioxidant response by impairing cystine import and depleting GSH
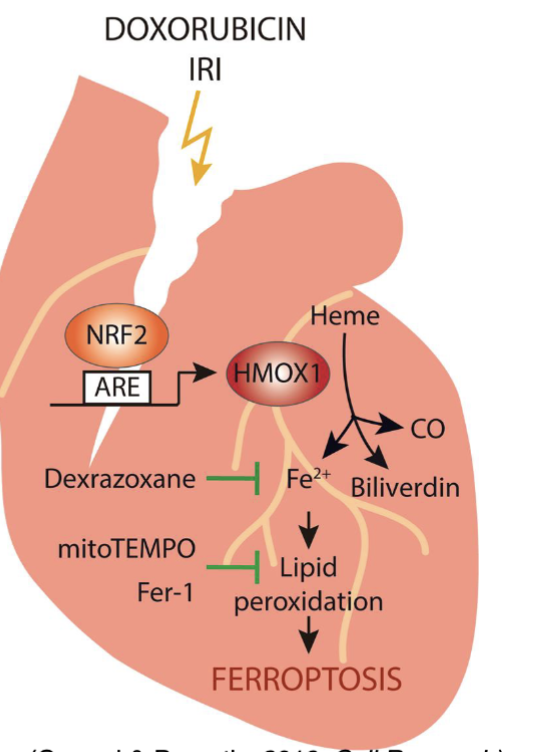
Doxorubicin also causes lipid peroxidation and depletes GSH
Does ferroptosis rely on DNA damage?
Ferroptosis is DNA damage independent
How can ferroptosis inhibition improve clinical outcomes in lung cancer therapy?
Ferroptosis inhibitor can reduce lung fibrosis after radiation therapy and reduce cardiac toxicity after doxorubicin treatment
FLASH radiation
a novel form of radiotherapy that delivers radiation at an ultra-high dose rate, completing a course of treatment in less than a second
What is one hypothesis which explains how FLASH therapy reduces damage to normal tissue?
Induction of lipid peroxidation and Fenton chemistry (possibly because tumour tissues have higher iron)