Organische Chemie
1/166
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
167 Terms
Atomorbitale
s Orbitale sind Rund, das 2s hat eine Kugelförmige Knotenfläche. p Orbitale sind Hantelförmig und haben eine Knotenfläche. Die beiden Lappen haben unterschiedliche Vorzeichen.
Pauli-Prinzip
Jedes AO kann max. 2 Elektronen haben, mit unterschiedlichem Spin.
Hundsche Regel
Bei mehreren entarteten AOs besetzen Elektronen zunächst die Orbitale einzeln und mit parallelem Spin.
Konstruktive Überlappung
Wenn die gleichen Vorzeichen überlappen. Es entstehen Sigma MOs, sie sind bindend
Destruktive Überlappung
Wenn die unterschiedlichen Vorzeichen überlappen. Es entstehen antibindende Orbitale, die die Bindung schwächen. (Sigma Stern und Pi stern)
Bindungsdissoziationsenergie
Die Energie die Frei wird, wenn zwei AOs ein MO bilden. Ist dies 0 wird die Bindung nicht entsthen, da immer Energie frei wird bei Bindungen.
Hybridisierung
Der Prozess, bei dem Atomorbitale kombiniert werden, um neue, gleichwertige Hybridorbitale zu bilden, die die chemischen Bindungen in Molekülen beschreiben.
sp3 haben einen Winkel von 109.5, sp2=120 und sp=180
Induktive Effekte
Die Effekte, die durch die Elektronendichteverteilung in einem Molekül verursacht werden, wenn eine Atombindung mit unterschiedlicher Elektronegativität besteht. Diese Effekte beeinflussen die Stabilität und Reaktivität von Molekülen.
Nomeklatur
Methan, Ethan, Propan, Butan, Pentan, Hexan, Heptan, Octan, Nonan, Decan
Aussehen Amid
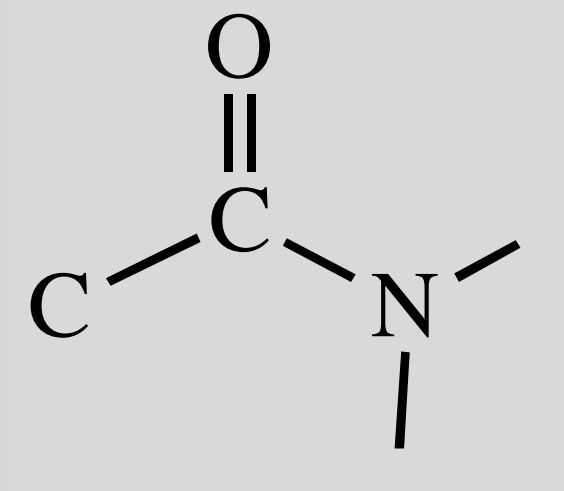
Nomenklatur: -amid
Aussehen Ester
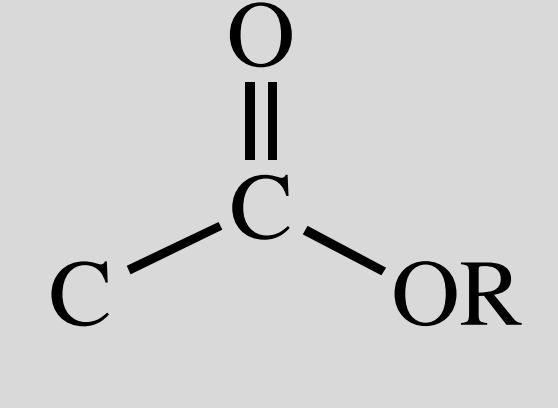
Nomenklatur: -oat
Aussehen Carbonsäure
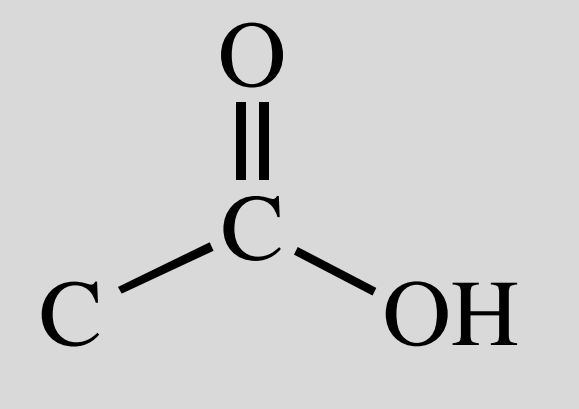
Nomenklatur: -säure
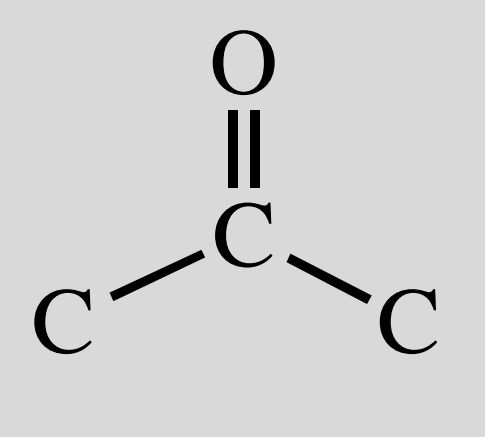
Keton
-on
Aldehyd
-al
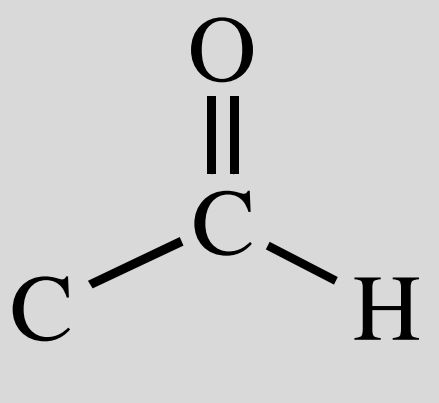
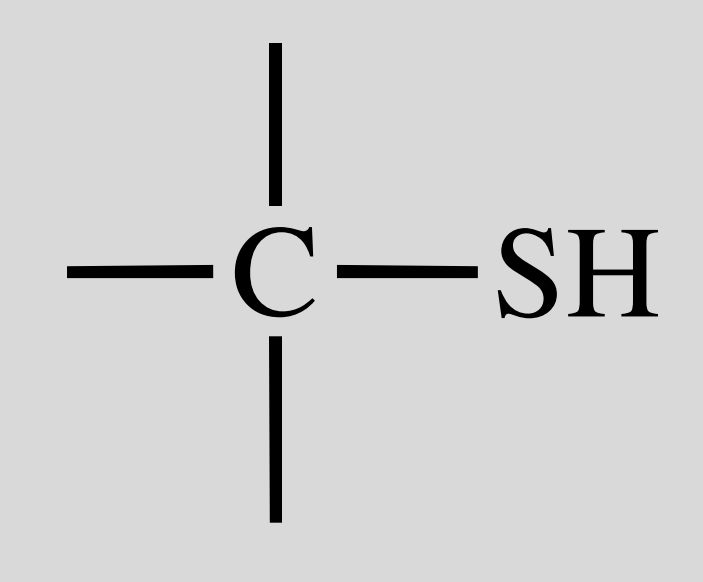
Thiol
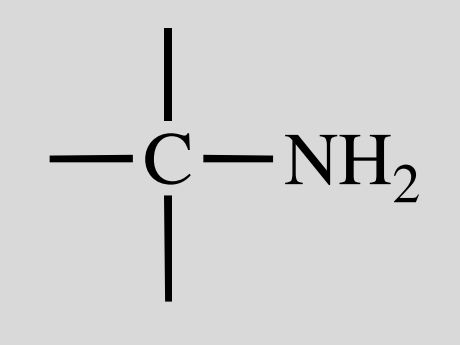
Amin
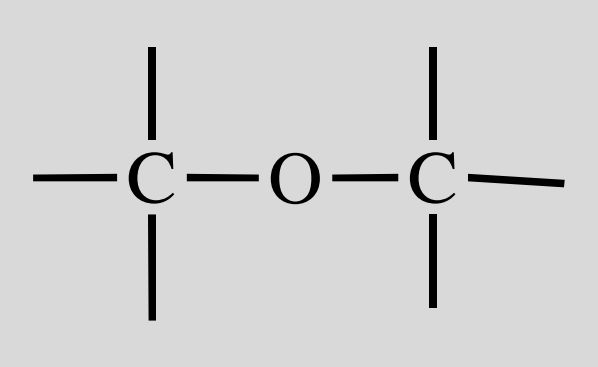
Ether
Halogenalkan
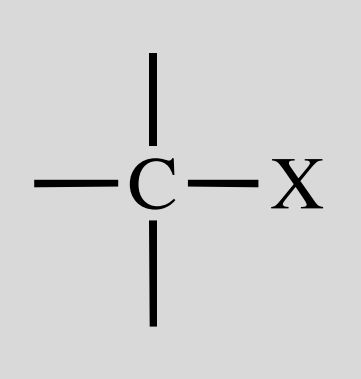
X= F,Cl,Br,I
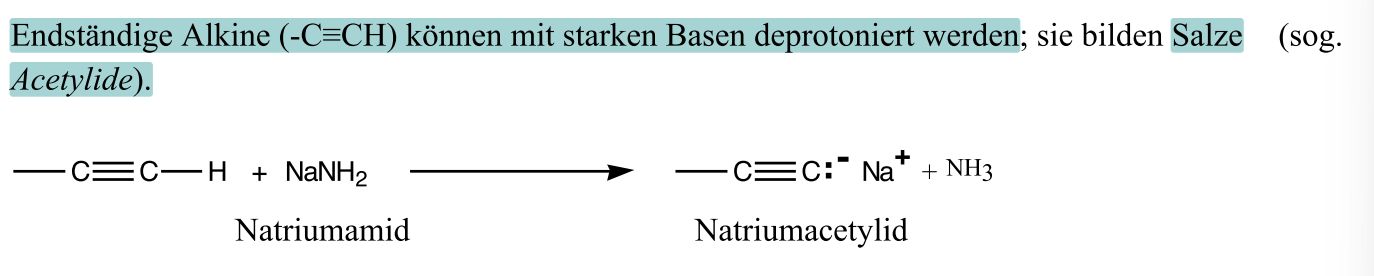
Salzbildung von Alkinen
Halogenwasserstoff-Addition an Alkine
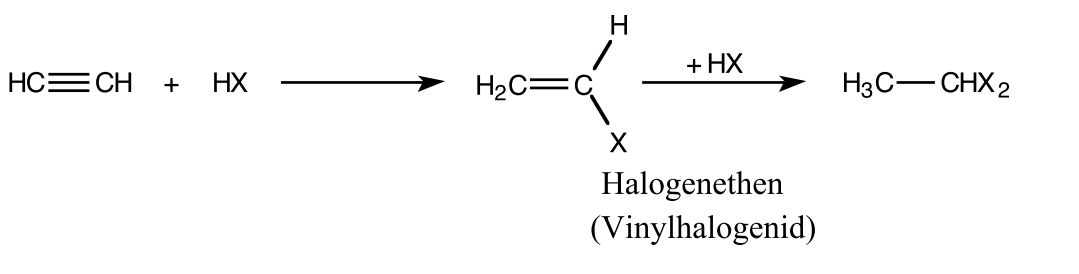
-> ionische Addition verläuft zweistufig
z.B. Herstellung von PVC
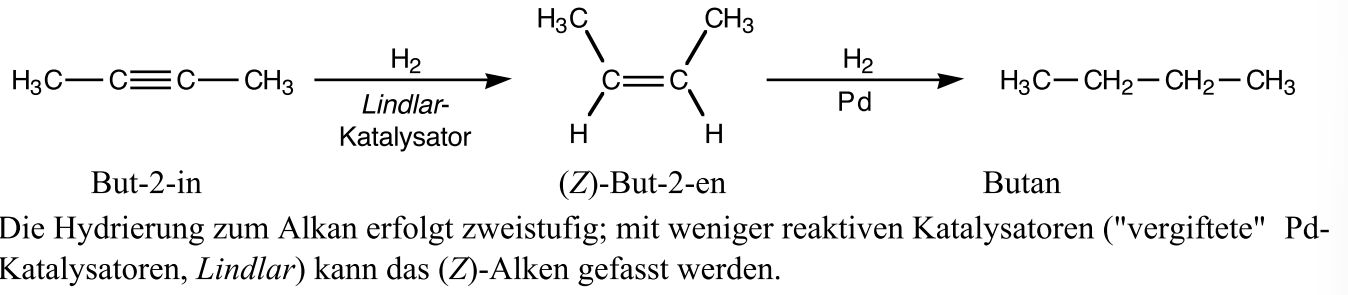
Katalytische Hydrierung von Alkinen (Butin)
Di- und Polyene mit kumulierten Doppelbindungen
Verbindungen mit gerader Anzahl kumulierter Doppelbindungen sind nicht planar gebaut --> Ebenen benachbarter pi-Systeme sind orthogonal zueinander
-> sie sind auch reaktiv und polymerisieren leicht

Isopren
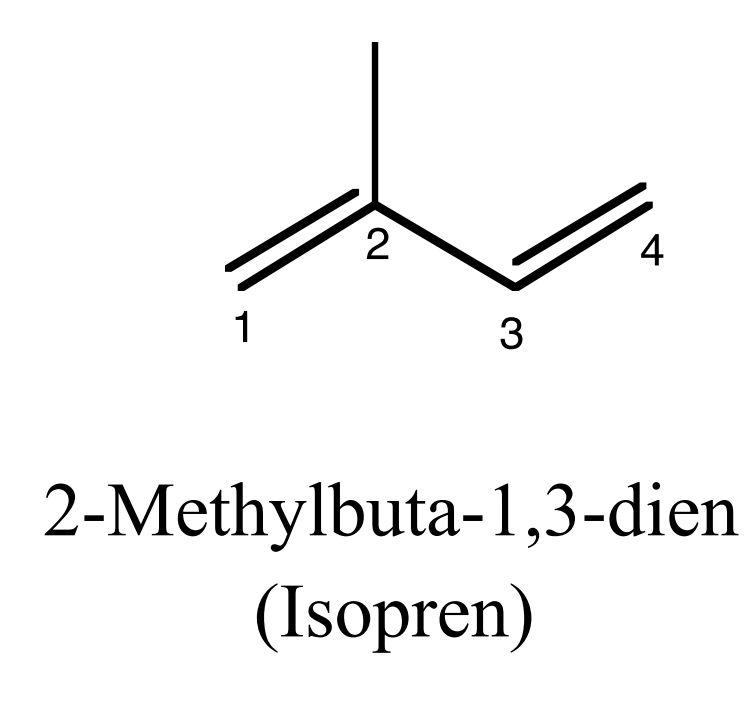
-> kommt als Baustein vieler Naturstoffe vor
z.B. beta-Carotin (Farbstoff Karotte) -> Vorstufe Vitamin A und 11-cis-Retinal, was wir zum Sehen brauchen
-> Polyene weisen Lichtabsorption in Abhängigkeit der Zahl der konjugierten C=C Doppelbindungen auf
Verschiedene Arten von Polyene
Polyene = Verbindungen mit mehreren C=C Doppelbindungen

-> jede C=C Einheit kann für sich betrachtet werden
-> C-Atome sind sp2 hybridisiert
Radikalische Polymerisation: Herstellung von Polypropylen (Additionsredaktion an Alkene)
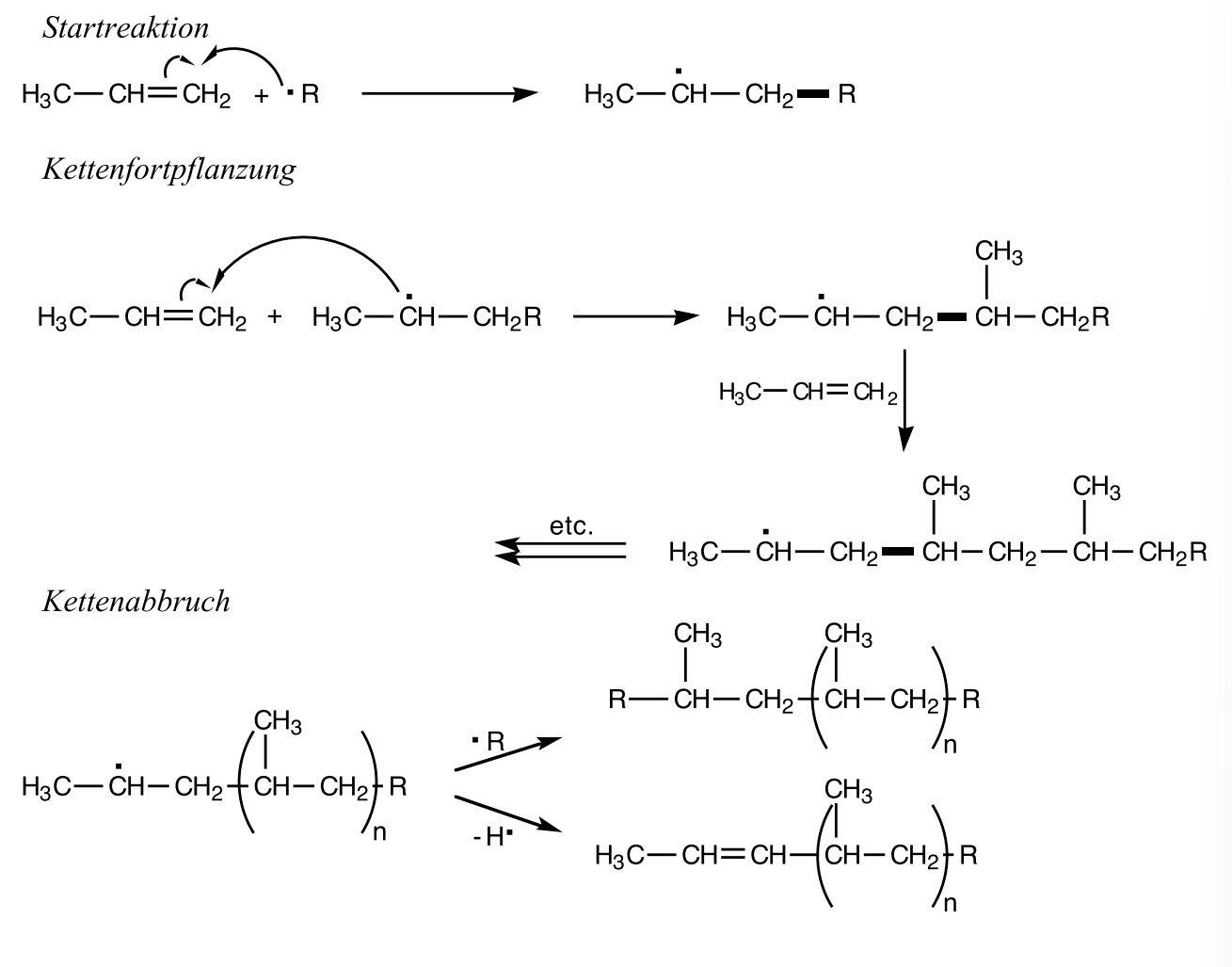
→ Polymere sind z.B. PVC, Teflon, Plexiglas
Epoxidierung (Additionsreaktionen an Alkene)
-> Oxirane (Epoxide) können durch die Reaktion von Alkenen mit Persäuren gewonnen werden

Katalytische Hydrierung (Addition an Alke
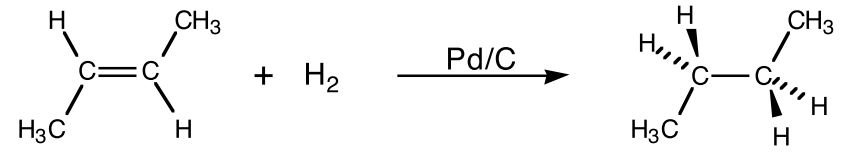
H2 wird an Metalloberfläche aktiviert und beide H-Atome addieren sich von der selben Seite an Doppelbindung => cis-Addition
Addition von Wasser an Alkene
Alken einer wässrigen Lösung einer Säure aussetzten:
Wasser ist abfangendes Nukleophil
pi-Elektronen der Doppelbindung greifen Proton an
Wasser (Nukleophil) bildet neue C-O sigma-Bindung, bindet also an positives C
Sauerstoff verliert ein Proton und es ergibt ein neutrales Molekül
in Gegenwart einer Säure liegt ein Glgw. zwischen Alkohol und Alkenen vor
-> zum Alken: Dehydratisierung
-> zum Alkohol: Hydratisierung
Halogen-Addition an Alkene
→ Halogene wie Chlor, Brom und Jod addieren an eine Doppelbindung und bilden dabei vicinale Dihalogenide.
z.B. Reaktion mit Brom
pi-Elektronen der Doppelbindung greifen das Brom an
die C-Br sigma-bindung wird gespalten
Bromium-Ion als Zwischenprodukt, Br- greift das Carbocation an
neutrales Endprodukt
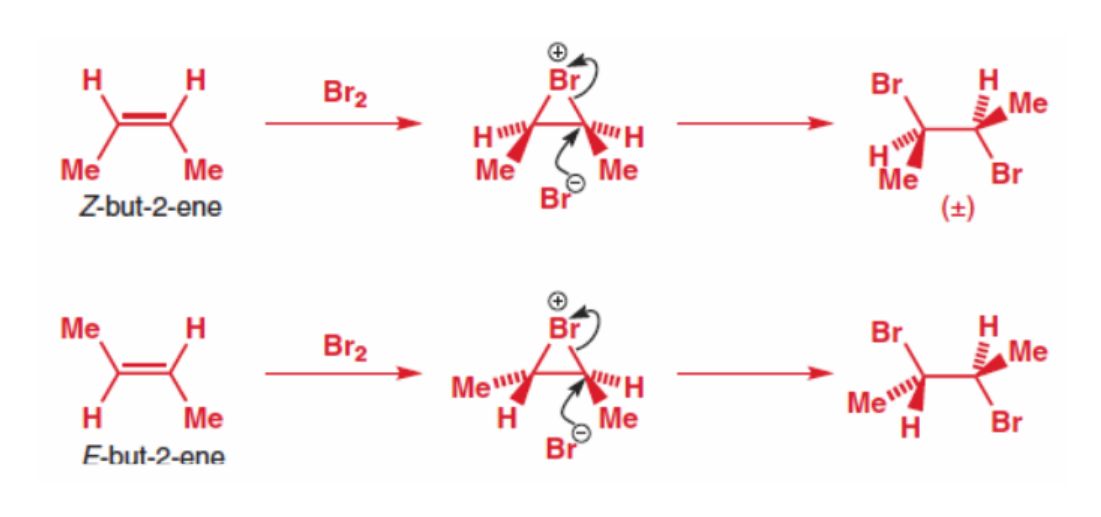
-> stellt eine anti-Addition dar: (alle C liegen auf Papierebene)

wenn aber die Bromaddition in einem nukleophilen Lösungsmittel (Methanol/ Wasser) durchgeführt wird, dann wird dieses an das stabiliere Kation addiert (pos. geladenes C) und nicht das Br-
1. Schritt, also Angriff von Brom auf Alken ist der langsame, geschwindigkeitsbestimmende Schritt
je elektronenreicher die Doppelbindung, desto schneller erfolgt der Angriff, da dann HOMO (Doppelbindung, erhöht) und LUMO (Br2) näher sind
Mesomerie
Verschiebung von pi-Elektronen führt zu mesomeren Strukturen
Hyperkonjugation
(sigma-Konjugation)
beruht auf Überlappung eines sigma-Orbital -> Alkylgruppe gilt als elektronenliefernd und kann benachbarte positive Ladungen stabilisieren
--> Stabilität von Carbenium-Ionen nimmt mit zunehmender Alkylsubstitution zu
--> Stabilität nicht nur wegen Hyperkonjugation, auch wegen induktiven (elektronenschiebenden) Effekten der Alkylgruppen, diese treten wegen der Polarisation also Elektronegativitätsunterschieden auf --> pos. Ladung teilweise kompensieren
Regel von Markovnikov (Addition von Halogenwasserstoffen HX an Alkene)
Wenn die an der Doppelbindung beteiligten Kohlenstoffatome nicht den gleichen Substitutionsgrad haben, addiert das Proton (elektrophil) des HX an das weniger substituierte Kohlenstoffatom. (regioselektiv)
--> für Alkene, die ähnlich substituiert sind, sind Produktgemische zu erwarten
Thermodynamik
Bestimmt ob Reaktion abläuft
Kinetik
bestimmt wie schnell Reaktion abläuft
Arrhenius Gleichung
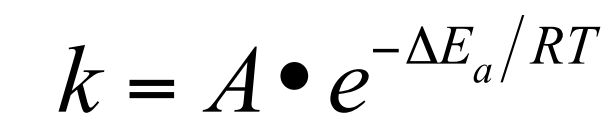
-> Geschwindigkeitskonstante k ist von der Temperatur abhängig
-> die Höhe der Aktivierungsenergie Ea bestimmt, wie schnell die Reaktion abläuft
-> langsamste Teilreaktion ist immer geschwindigkeitsbestimmend
Gleichgewichtskonstante K
Keq = Konz. Produkte/ Konz. Edukte

ΔG Gibbsche freie Energie
entspricht der Energiedifferenz zwischen Produkten und Edukten und bestimmt, ob eine Rkt. freiwillig ablaufen kann oder nicht
--> muss negativ sein, damit Rkt. spontan abläuft

S = Entropie (Änderung der Ordnung des Systems)
H = freie Enthalpie
Anhydro-
Wasserabspaltung
Des-
Fehlen einer funktionellen Gruppe und Ersatz durch H
Cyclo-
Ringschluss zwischen zwi zu bezeichnenden C-Atomen
Dehydro-
an anzugebender Prosition dehydriert
Homo-
Mehrgehalt von CH2
Nor-
Fehlen von CH3, CH2 oder CH
Konstitutionsisomerie
besitzen gleiche Summenformel aber unterscheiden sich durch Verknüpfungsweisen ihrer Atome und Gruppen (Konstitutionen= Verknüpfungen)
kovalente Bindung
Atome teilen Elektronen miteinander, jedes Atom erhält so formal ein äusseres Elektronenoktett
Ionenbindung
elektronegatives Atom (Tendes Elektron an sich zu ziehen) + elektropositives Atom (Tendenz Elektronen zu übertragen)
Substitutionsreaktion
= eine funktionelle Gruppe (Abgangsgruppe) in einem Molekül wird durch eine andere ersetzt
—> Anzahl Eduktmoleküle=Anzahl Produktmoleküle
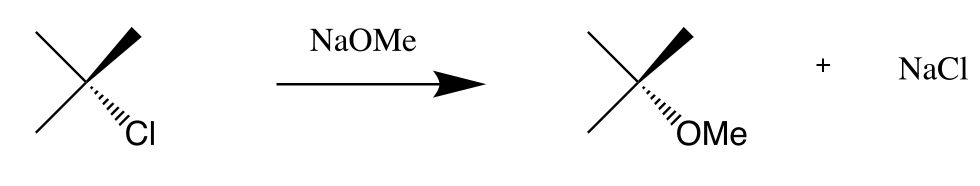
Additionsreaktion
= ein Molekül wird an ein zweites addiert
--> Anzahl Edukt-Moleküle ist grösser als Anzahl Produkt-Moleküle

Reaktionsmechanismus
= Art und Weise, wie Elektronen übertragen werden
--> Bewegung der Elektronen wird durch gebogene Pfeile dargestellt, Pfeilspitze gibt Zielort des Elektrons an

Elektophil
- elektronenarmes Atom
- nimmt e- auf
- haben positive Ladung, ein leeres p-Orbital, eine Doppelbindung zu einem elektronegativen Atom oder eine Einfachbindung zu einem elektronegativen Element

Nukleophil
- elektronenreiches Atom
- gibt Elektronen ab
- haben entweder einsames Elektronenpaar, negative Ladung, elektronenreiche Mehrfachbindung oder besitzen eine Bindung zu einem elektropositiven Atom

Bindungsneubildung von Kovalenzbindung auf drei vers. Arten:
1. Radikalrekombination
2. Ladungskomplementierung
3. Reaktion zwischen ungeladenen Elektrophilen und Nukleophilen

radikalische Substitution
mehrere Schritte:
1. homolytische Spaltung eines Chlormoleküls --> Chlorradikal wird gebildet
2. Chlorradikal reagiert mit Alkan unter Abstraktion eines Wasserstoffradikals unter Bildung des Halogenwasserstoffs, wobei Alkylradikal gebildet wird
3. dieses kann mit weiterem Chlormolekül zum Halogemalkan (unreaktiv) reagieren

mehrkernige benzoide Kohlenwasserstoffe
=> durch Kondensation oder Annelierung mehrerer Benzolringe
an diesen Ringen kann man elektrophile Substitutionen durchführen, wie an Benzol
Kontrolle der Regioselektivität ist hier schwieriger
viele davon sind karzinogen (krebserregend)
z.B. Benz(a)pyren -> entsteht bei Verbrennung organischer Materie (Autotreibstoff, Erdöl, Müllverbrennung, Waldbränden, Zigaretten, gegrilltem Fleisch, heissen Kochen)
-> Enzym oxidiert Pyren in Epoxid, Basenstruktur in DNA verändert durch Epoxid , Mutation, wuchernde Zellen = Krebs
Synthese von Benzol(derivaten)
bei der Synthese eines spezifischen Benzolderivats, muss der erste Substituent alle weiteren Substituenten an die richtige Position dirigieren
bestimmte Rkt. können dirigierende Wirkung eines Substituenten umkehren
z.B. Alkyl Seitenkette zu Carbonsäure oxidieren
z.B. meta-dirigierende Nitrogruppe in ortho/para-dirigierende Aminogruppe --> Nitrogruppen in Aminogruppen reduzieren
(Friedel-Crafts Rkt sind an Nitrobenzol nicht möglich, da es zu stark desaktivierend wirkt)
elektrophiler Angriff auf disubstituierte Benzole
Wirkungen zweier Substituenten (auf die rel. Gesw. und Orientierung der Substitution) addieren sich
wenn Substituenten um selbe Stelle konkurieren, setzt sich die stärker de-/aktivierende Gruppe durch -> Resonanz Effekte sind normalerweise stärker als Induktive Effekte
Stellung zwischen räumlich ausgedehnten Gruppen ist aus sterischen Gründen oft ungünstig
in den meisten anderen Fällen entstehen Produktgemische
Friedel-Crafts Alkylierung
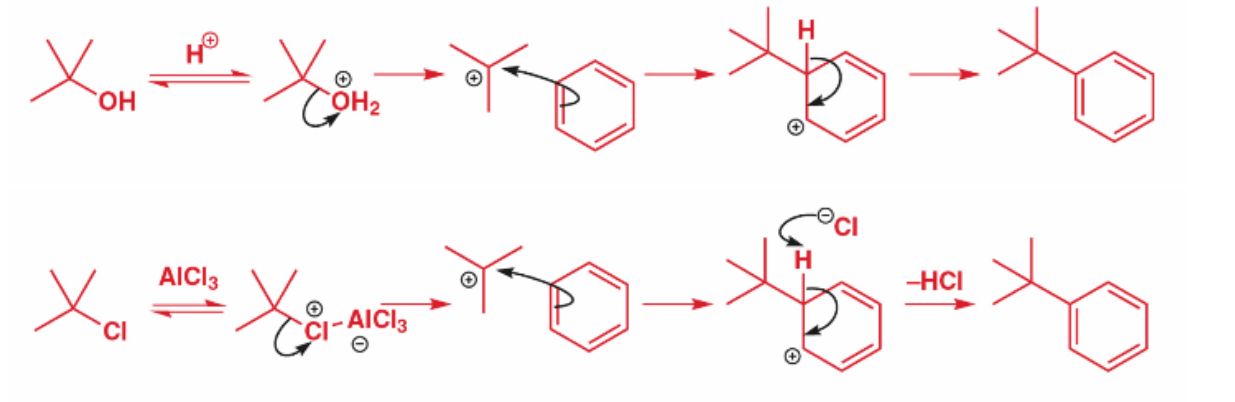
1. Alkylkation wird aus Alkylhalogenid durch eine Lewis Säure oder durch Protonierung mit Wasserabspaltung gebildet
2. Carbokation greift als Elektrophil den Aromaten an (H+ abgespalten)
typische Lewis-Säuren nach abnehmender Reaktivität: AlBr3, AlCl3, FeCl3, SbCl5 und BF3
Probleme:
Produkt ist reaktiver als Edukt -> mehrfach Alkylierungen
Umlagerungen des Carbokations -> statt dessen Friedel-Crafts Acylierung verwenden
Sulfonierung des Benzolrings
"Rauchende Schwefelsäure" Oleum protoniert sich selbst und bildet das Sulfonium Ion:

Nitrierung des Benzolrings
Nitriersäure = Salpetersäure (HNO3) + Schwefelsäure (H2SO4)


1. Schwefelsäure protoniert als stärkere Säure die Salpetersäure
2. die protonierte Salpetersäure spaltet Wasser ab und das Nitronium Ion entsteht, das positiv geladen und somit elektrophil ist
3. das Elektrophil greift dann den Aromaten an (H+ wird wieder abgespalten)
Elektrophile aromatische Substitution
-> Substitution eines H-Atoms


-> der 1. Schritt ist th'dynamisch ungünstig, da die Aromatizität verloren geht, aber danach wird der aromatische Ring wieder regeneriert, indem das H+ abgespalten wird
--> Bildung des ersten Übergangszustand ist geschwindigkeitsbestimmend, da er langsamer ist
--> diese Rkt. ist energetisch günstiger als Rkt. mit einem Nukleophil, dabei würde ein Additionsprodukt entstehen
Hydrierungswärmen
Mass für die relative Stabilität von Alkenen anhand Hydrierungswärmen
Unterschied zwischen Hydrieungswärmmen=Resonanzenergie
andere Namen: Delokalisierungsenergie, aromatische Stabilisierung, Aromatizität
sigma-Skelett
6 sp2-hybridisierte C-Atome liegen in der Ebene
Überlappung von je 2 hybridisierten Orbitalen bilden 6 sigma-Bindungen --> Kohlenstoff-Sechsring und 6 C-H Bindungen
pz-Orbitale sind je mit 1 e- besetzt und stehen senkrecht zur Ringebene
pi Elektronen überlappen seitlich und bilden pi-MOs --> energetisch tiefstes MO, wenn alle pz Orbitallapen Vorzeichen auf gleicher Seite haben
Stellungen der Substituenten in disubstituierten Benzolderivaten
ortho: 1,2-
meta: 1, 3-
para: 1, 4-
Substituenten in alphabetischer Reihenfolge nennen
ipso: ein bestehender aromatischer Substituent wird in Reaktion gegen einen anderen ausgetauscht

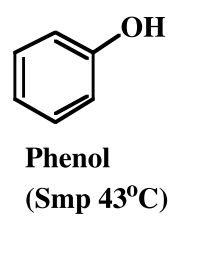
Phenol
Toluol
Wie heisst dieses heterocyclische Aromat?
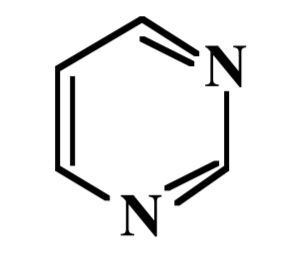
Pyrimidin
Wie heisst dieses heterocyclische Aromat?
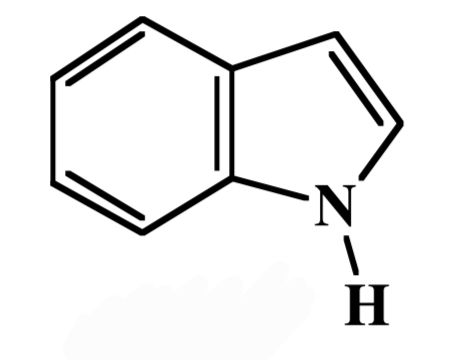
Indol
Wie heisst dieses heterocyclische Aromat?
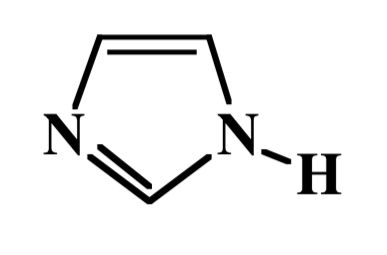
Imidazol
Wie heisst dieses heterocyclische Aromat?
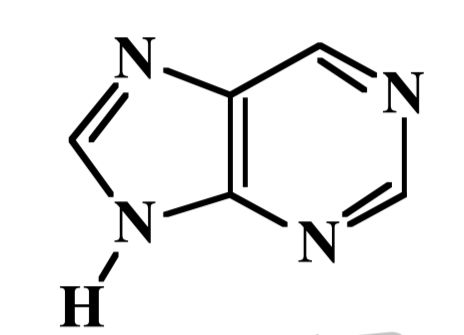
Purin
Wie heisst dieses heterocyclische Aromat?
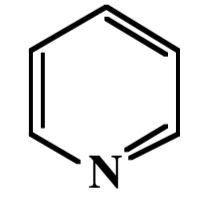
Pyridin
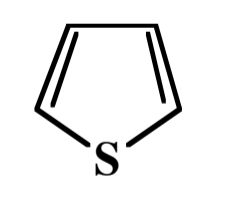
Wie heisst dieses heterocyclische Aromat?
Thiophen
Wie heisst dieses heterocyclische Aromat?
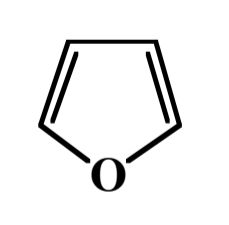
Furan
Wie heisst dieser heterocyclische Aromat?
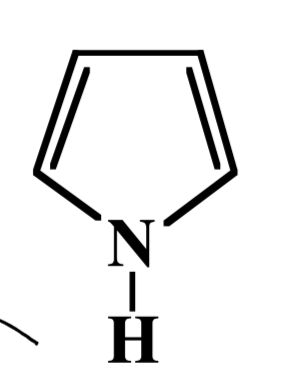
Pyrrol
Was ist das für ein Heterocyclen, wie entsteht es und was hat es für Eigenschaften?

Imidazol (aromatisches Heterocyclen)
entsteht wenn man ein weiteres CH im Pyrrol durch N ersetzt (-> es gibt noch weitere)
kommt in Seitenkette der AS Histidin vor, biologisch wichtig
bei der Decarboxylierung von Histidin entsteht Histamin --> bildet sich in allen Geweben, viel freies Histamin ist Ursache vieler Allergien
beide N im Imidazol können protoniert und deprotoniert werden -> beim physiologischen pH (~ 7) ist die Hälfte protoniert, die andere deprotoniert

Wie kann man Enantiomere trennen?
wenn man eine Verbindung in einer enantiomerenreinen Form erhalten will
1. nur ein Enantiomer synthetisieren (enantioselektive Synthese)
2. Racematspaltung: aus dem Racemat die Enantiomere voneinander trennenEnantiomere lassen sich schwer trennen, da sie identische Eigenschaften besitzen, Diastereomere besitzen jedoch unterschiedliche Eigenschaften und man kann sie durch übliche Trennverfahren trennen (fraktionierte Kristallisation, Destillation, Chromatographie) --> deshalb wird ein Enantiomergemisch zuerst in ein Diastereomerenpaar überführt (durch Salz/ enantiomere Hilfsgruppe)

Was bedeutet mehr als 2 Chiralitätszentren zu haben?
allg. gilt; eine Verbindung mit n Chiralitätszentren kann maximal 2n Stereoisomere haben (Permutation)
z.B. 3 Chiralitätszentren = 8 Stereoisomere
Zahl der möglichen Stereoisomere nimmt ab, wenn Mesoformen und gewisse cyclische Systeme auftreten
Was sind Meso-Verbindungen?
= wenn eine Verbindung zwei (oder mehr) Chiralitätszentren enthält, aber deckungsgleich mit ihrem Spiegelbild ist, lassen sich durch Drehung zur Deckung bringen
Mesoverbindungen besitzen eine Spiegelebene, die das Chiralitätszentrum auf das andere abbildet
sind also achiral und deswegen optisch inaktiv
z.B. Weinsäure:

=> Verbindungen mit mehreren Stereozentren können, müssen aber nicht, chiral sein
Was sind Diasteromere?
in Molekülen mit mehreren Chiralitätszentren, sind mehrere Stereoisomere möglich
Chiralitätszentren entweder R oder S konfiguriert
z.B. AS Threonin mit 4 Stereoisomeren, und 2 Enantiomerenpaare


Diastereomere = Stereoisomere, die sich nicht wie Bild und Spiegelbild verhalten
Diastereomere unterscheiden sich in ihrer Struktur (Geometrie), also sicher in Bindungslänge, Bindungswinkel oder Torsionswinkel
also haben sie unterschiedliche Energiegehalte, und deshalb auch unterschiedliche physikalische und chemische Eigenschaften (nicht wie Enantiomere)
alle Stereozentren im Molekül werden invertiert --> erhält man das Enantiomer, wenn nicht alle Zentren invertiert werden --> erhält man ein Diastereomer

Was sind verschiedene Darstellungsformen von Sturkturformeln?
Newman Projektion: um C-C Einfachbindung rotieren, günstigste Konf. ist die gestaffelte Anordnung, grösste Substituenten sind anti-periplanar angeordnet
Sägebockprojektion
normale Darstellungsform: C-Kette als horizontale Zickzacklinie in Papierebene, Substituenten sind dann hinter oder vor Papierebene
Fischerprojektion: horizontale Substituenten zeigen aus Papierebene nach vorn --> muss um 180° gedreht werden, um in gestaffelte Form zu kommen
Was ist die Fischer-Projektion?
Standardmethode zur zweidimensionalen Abbildung tetraedischer Kohlenstoffatome und ihrer Substituetnten
Kreuz, mit chiralem C in der Mitte
waagrechte Linien stellen Bindungen dar, die zum Betrachter gerichtet sind
senkrechte Linien weisen vom Betrachter weg

Was sind die D und L Konfiguration?
vor der Röntgenstrukturanalyse war die absolute Konf. chiraler Moleküle unbekannt
willkürlich wurden den beiden Enantiomeren vom 2,3 Dihydroxypropanal (lässt sich in viele andere chirale Moleküle führen) bestimmte Konfigurationen zugeordnet
Konf. bezieht sich nicht auf das Vorzeichen der Drehung des linear polarisierten Lichts, sondern auf die relative Anordnung der Substituenten
rechtsdrehend: D (dexter, rechts)
linkdrehend: L (laevulus, links)

Schreibweise: 1. D/L-Konf., 2. Drehsinn (+)/(-) in Klammern, 3. Name der Verbindung
wird noch bei Zuckern und AS verwendet, aber stollte durch R/S-Konf. ersetzt werden
Was sind anomale Dispersionen?
= bestimmte Form der Röntgenstrukturanalyse
Bestimmung ob positives oder negatives Vorzeichen
ermittelt neben Struktur auch die absolute Konfiguration
Was ist das Vorgehen des CIP-Nomenklatursystem?
= System, um Händigkeit von Molekül zu bestimmen, Enantiomere zu benennen
1. Substituenten nach abnehmender Priorität ordnen (1-4)
2. Molekül so drehen, dass Substituent mit geringster Priorität nach hinten zeigt
3. Pfeil von 1 über 2 nach 3
-> wenn gegen Uhrzeigersinn, dann S Konf. (sinister)
-> wenn im Uhrzeigersinn, dann R Konf. (rectus)
z.B. (S)-alanine
in was unterscheiden sich zwei Enantiomere?
in ihren Topographien
sie haben verschiedene räumliche Anordnungen der Substituetnten um das Chiralitätszentrum
unterschiedliche Händigkeiten
unterschiedliche absolute Konfiguration
keine Beziehung zwischen Vorzeichen des Drehwerts und der absoluten Konfiguration
Unterscheiden durch:
Polarimeter: wenn man einen Strahl von linear polarisiertem Licht (chiral) durch eine Probe der Enantiomere schickt
die Schwingungsebene des einfallenden Lichtes wird um einen bestimmten Betrag in eine Richtung gedreht, also im oder gegen den Uhrzeigersinn
die Schwingungsebene wird bei beiden Enantiomeren um denselben Betrag aber in die entgegengesetzte Richtung gedreht => optische Drehung, Probe ist optisch aktiv

rechtsdrehend (dextrorotatory) = Enantiomer, das Ebene des polarisierten Lichts im Uhrzeigersinn dreht -> (+)-Enantiomer
linksdrehend (levorotatory) = Enantiomer, das Ebene des polarisierten Lichts gegen den Uhrzeigersinn dreht -> (-)-Enantiomer
bei achiralen Molekülen bleibt die Richtung unverändert --> optisch inaktiv
Was ist ein Racemat und eine Racemisierung?
Racemat/ racemisches Gemisch = ein 1:1 Gemisch von Enantiomeren
Racemisiserung = wenn ein Enantiomer über irgendeinen Prozess mit seinem Spiegelbild ins Gleichgewicht gebracht wird, kann durch Konformationsänderung erfolgen
Was ist die Röntgenstrukturanalyse?
=Methode zur Aufklärung der Struktur eines Moleküls im kristallinen Zustand
-> nach Bestimmung der Positionen der Atome, kann man Bindungslängen und -Winkel und andere geometrische Faktoren ableien
Was ist Methylierung?
= nukleophile Substitutionsreaktion an sp3 Zentren als Reaktion in biologischen Systemen
Überführung einer Methylgruppe von einem Elektrophil an ein Nukleophil
im Labor Methyljodid dafür verwenden
in Natur S-Adenosylmethionin SAM
z.B. im Gehirn Adrenalin aus Noradrenalin aufbauen
z.B. Methyltransferasen methylieren Basen in der DNA
Was bestimmt der Grad der Substitutio des reaktiven Kohlenstoffatoms?
-> ob Halogenalkane (oder ähnliche Derivate) mit Nukleophilen bevorzugt nach SN1 oder SN2 reagieren:
3°-Halogenalkane nach SN1-Mechanismus -> besondere Stabilität, Dissoziation in Ionen erfolgt leicht
1°-Halogenalkane nach SN2
2°-Halogenalkane meist nach SN2, wenn Carbeniumionen stabilisiert sind, dann auch SN1 möglich

Was ist die Allylstellung?

pi-Elektronen können verschoben werden -> Elektronendelokalisierung
zwei äquivalente mesomere Grenzformeln bilden
mesomere Teilchen sind besonders stabil und lassen sich gut bilden
es kann dabei zu Produktgemischen kommen (beide/ mehrere Grenzstrukturen kommen vor -> Stabilisierung)
Wie nimmt die Stabilität von Carbenium-Ion zu?
in der Reihe 1° < 2° < 3°
nimmt mit zunehmender Alkylsubstitution zu

hängt mit +I Effekt von Alkylsubstituenten und Hyperkonjugation zusammen:

Was für ein Einfluss haben sterisch gehinderte Halogenalkane auf die Geschwindigkeit der SN2 Reaktion?
sterisch gehinderte Halogenalkane reagieren nur sehr langsam, da das nukleophile Zentrum immer mehr sterisch abgeschirmt ist, je höher der C substituiert ist:

Wie kann die OH Gruppe doch zu einer guten Abgangsgruppe werden?
1. durch Überfühlrung in ein Sulfonat wird OH Gruppe zu guter Abgangsgruppe

2. durch Protonierung des freien Elektronenpaars des O Atoms erhält man ein Oxonium-Iom H3O+ (gute Säure), aus OH wird Wasser, eine gute Abgangsgruppe

Eliminierungen vs. nukleophile Substitutionen
Grösse Nukleophil/ Base:
Eliminierung: Angriff des Nukleophils am Proton des benachbarten C
Substitution: Angriff des Nukleophils am C, der den Substituenten trägt
-> H ist besser zugänglich als C, deshalb greifen sterisch anspruchsvolle Basen an der alpha-Position an und führen zur Eliminierung
-> für Eliminierungen also sterisch anspruchsvolle, nicht nukleophile Basen benutzen (z.B. tert-Butyloxid, Stickstoff Base DBU)
Basizität:
starke Basen greifen eher H an -> Eliminationsreaktionen
schwache Basen greifen H eher nicht an -> Substitution
Temperatur:
hohe Temperatur begünstigt Eliminierung wegen Th'dynamik:
#Produkt-Teilchen > #Edukt-Teilchen -> Entropie-begünstigt und lauft deshalb bei höherer Temperatur besser
Übersicht E1, E2, E1eB
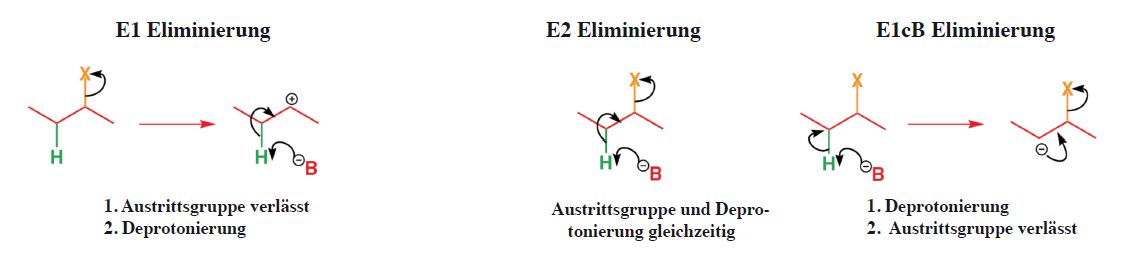
Durch was wird eine Eliminierung begünstigt?
hohe Temperatur:
#Produkt-Teilchen > #Edukt-Teilchen -> Entropie-begünstigt und lauft deshalb bei höherer Temperatur bessergrosse Basizität
sterisch anspruchsvolle Basen
Eliminationsreaktion
Halogenalkane können ausser Sustitutionsreaktionen auch durch Eliminierung mit Nukleophilen reagieren
dies, weil Nukleophile oft gute Basen sind.
Reaktionsprodukt enthält eine Doppelbindung
nützlich für Herstellung von Alkenen
Wie sieht die Oxidation von Alkoholen aus?
Oxidation von Alkoholen zu Aldehyden, Ketonen oder Carbonsäuren.
primäre Alkohole → Aldehyden/Carbonsäuren
sekundäre Alkohole → ketone
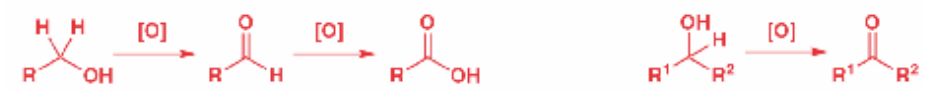
- häufig verwendetes Reagenz zur Oxidation ist Chrom
- eine Weiterreaktion zur Säure lässt sich vermeiden, wenn man wasserfrei arbeitet
Bildung von Halogenalkanen
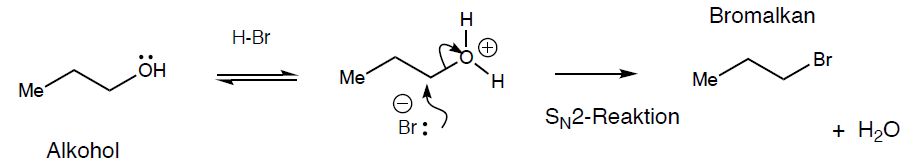
- entstehendes primäres Carbenium-Ion ist energiereich & instabil und können von einem Nukleophil angegriffen werden
- 2° & 3° Alkohole lassen sich durch HCl/ HBr in Halogenalkane umwandeln = SN1
Dehydratisierung?
durch Protonierung von Alkoholen entstehen Oxonium-Ionen
sekundäre und tertiäre Oxonium-Ionen können daraufhin Wasser abspalten und stabile sekundäre/ tertiäre Carbenium-Ionen ausbilden
es folgt ein Wasserverlust nach einem E1 Mechanismus
das Carbenium-Ion verliert ein Proton und ergibt ein Alken

Wie wird Ether aus Alkoholen hergestellt?
durch Reaktion eines Alkoxids mit einem primären Halogenalkan oder einem Sulfonsäureester (Mesylat/ Tosylat) unter SN2 Bedingungen
= Williamson-Ethersynthese
Alkoxide: Alkohol + Na -> starke Basen, nur bei primären ungehinderten Systemen
