Chapter 7- Three Dimensional Structure of Proteins
1/65
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
66 Terms
4 Core Pauling Principles
Protein Secondary Structures must meet rules of:
Bond lengths and angles must stay close to those observed by X-ray crystallography.
Steric limits: Atoms cannot come closer than van der Waals radii allow, so not all bond rotations are possible.
Planarity of peptide bond: The peptide bond is rigid and planar; rotation happens only around the ϕ (N–Cα) and ψ (Cα–C) bonds.
Stability via H-bonds: Patterns must be stabilized by hydrogen bonding between backbone C=O oxygens and N–H protons.
This reduces possible shapes to a few stable conformations → mainly α helix and β sheet.
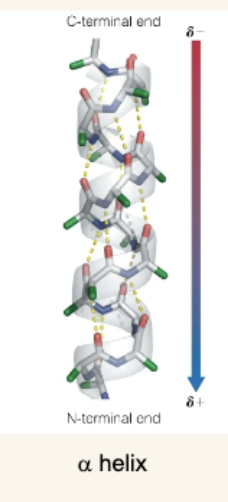
Components of the α Helix
Right-handed helix with 3.6 residues per turn.
Pitch (one full turn) = 5.4 Å; Rise per residue = 1.5 Å.
Stabilized by H-bonds between residue i carbonyl oxygen and i+4 amide hydrogen.
Has a macrodipole: N-terminus is partially positive, C-terminus partially negative.
Side chains radiate outward.
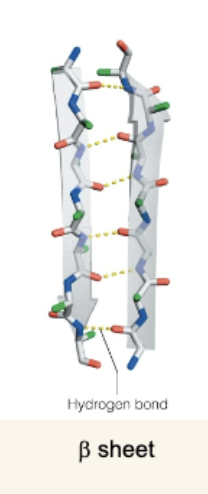
The β Sheet
Made of β strands connected by H-bonds.
Two orientations:
Antiparallel: N→C in one strand runs opposite the neighbor (stronger H-bonds).
Parallel: N→C in same direction; H-bonds angled and less stable.
Side chains alternate above and below the sheet → can be amphipathic.
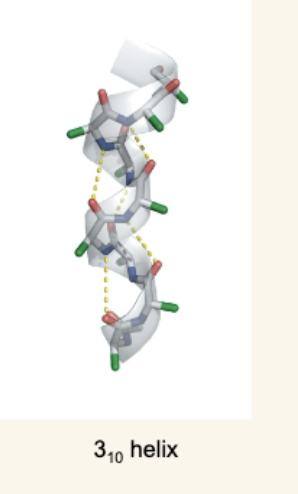
Less Common Helices
3₁₀ helix: Tighter than α-helix, H-bonding between i and i+3 residues, less stable.
Polypeptide II helix (polyproline II): Left-handed, no backbone H-bonding, frequently has proline and glycine.
![<ul><li><p class="my-2 [&+p]:mt-4 [&_strong:has(+br)]:inline-block [&_strong:has(+br)]:pb-2"><strong>3₁₀ helix</strong>: Tighter than α-helix, H-bonding between i and i+3 residues, less stable.</p></li></ul><ul><li><p class="my-2 [&+p]:mt-4 [&_strong:has(+br)]:inline-block [&_strong:has(+br)]:pb-2"><strong>Polypeptide II helix (polyproline II)</strong>: Left-handed, no backbone H-bonding, frequently has proline and glycine.</p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/59e26089-8d0e-466a-86b6-683e96b49ab4.png)
Amphipathic Helices and Sheets
Secondary structures often organize so one face is hydrophobic (faces protein interior or other hydrophobic surfaces) and one face is hydrophilic (faces solvent).
α helix: Similar polarity side chains appear every 3–4 residues.
β sheet: Alternating polar/nonpolar residues create two distinct surfaces.
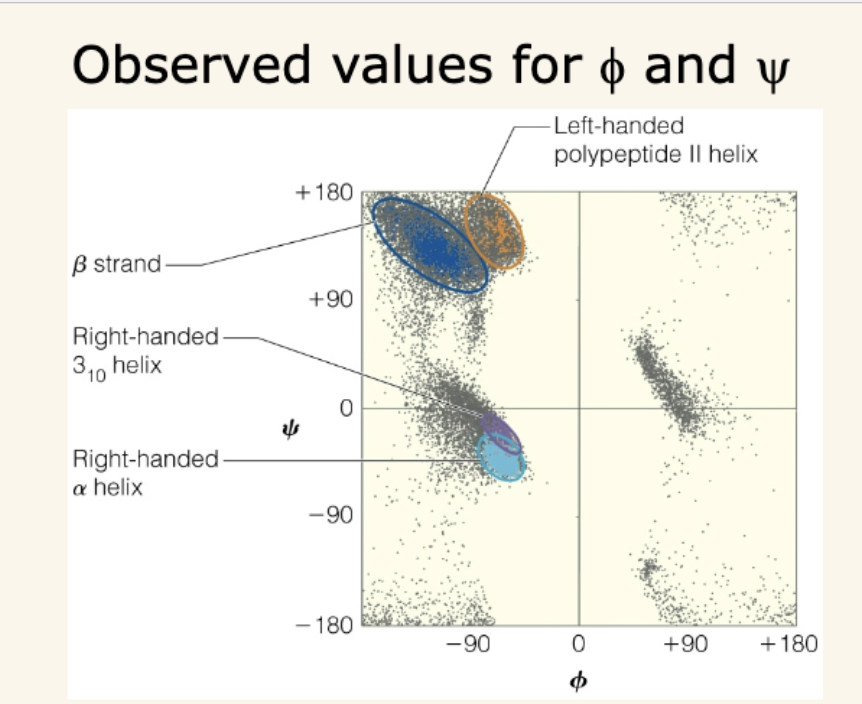
Ramachandran Plots
Show allowed ϕ and ψ angles, which describe polypeptide backbone conformation.
Steric hindrance eliminates most possible combinations.
Clusters indicate where α helices, β sheets, and other motifs lie.
Residue-specific effects:
Glycine → most flexible, more possible regions.
Proline → most restricted (can’t rotate around ϕ).
Key Table of Secondary Structures (simplified)
Structure | Residues/turn | H-bonding pattern | Notes |
|---|---|---|---|
α helix | 3.6 | i → i+4 | Common, right-handed, macrodipole |
3₁₀ helix | 3.0 | i → i+3 | Tighter, less common |
β sheet | varies | Between strands | Parallel or antiparallel |
Polyproline II | ~3.0 | None (main-chain) | Left-handed, enriched in Pro/Gly |
3 main types of fibrous proteins
α-Keratin
β-Keratin and Fibroin
Collagen
α-Keratin structure
Predominantly α-helical, with pairs of α helices twisted into a left-handed coiled coil.
α-Keratin location
Hair, skin, fingernails.
α-Keratin stability
Stabilized by hydrophobic stripes (caused by nonpolar side chains every 3-4 residues) and disulfide bonds (crosslinks especially in nails and hair for strength).
α-Keratin mechanical properties
Flexible or hard, depending on crosslinking
β-Keratin
Found in reptiles and birds (e.g., feathers), rich in β-sheet structure.
Fibroin
Main protein in silk, composed of stacked antiparallel β sheets.
β-Keratin and Fibroin Amino Acids
Enriched in glycine, alanine, and serine (allows tight sheet packing).
β-Keratin and Fibroin Properties
Strong and not stretchy, but flexible because the sheets are held by weak van der Waals forces, not covalent bonds.
Collagen Structure
Three left-handed helical polypeptide chains twisted into a right-handed triple helix.
Collagen Amino Acids
Repeats of Gly–X–Y, where X/Y often proline or hydroxyproline. Every third residue is glycine (smallest side chain, fits in the tight center).
Collagen Role
Major structural protein in bones, tendons, skin.
Collagen Stability
Hydrogen bonds and covalent crosslinks (via modified lysine).
Collagen Special Note:
Vitamin C is needed for proline/lysine hydroxylation. Deficiency leads to scurvy due to weakened collagen
Compact 3D Folding of Globular Proteins
Globular proteins fold their secondary structural elements (α helices, β sheets) into compact globular shapes.
The tertiary structure results from the packing of these secondary elements, making the protein functional.
Example: Ubiquitin (small globular protein with 76 residues) shows complex folding despite its size.
3 types of Structural Representations of Globular Proteins
Cartoon model
Stick Model
Surface Model
Cartoon model:
Shows the backbone and secondary structure (helices as ribbons, β strands as arrows).
Surface model:
Illustrates protein "skin"—solvent-accessible surface indicating hydrophobic (nonpolar) and hydrophilic (polar/charged) regions.
Hydrophobic residues tend to be buried inside; hydrophilic residues are exposed on the surface interacting with water/environment.
4 types of Diversity of Folding Patterns
Proteins can be broadly classified by secondary structure dominance:
Mainly α helix (e.g., myoglobin)
Mainly β sheet (e.g., neuraminidase)
Both α and β (e.g., triosephosphate isomerase, TIM)
Little α or β
Protein Domains
Compact, locally folded regions (150–250 amino acids).
Multiple domains in large proteins, each can have distinct functions.
Domains can contain supersecondary motifs like β/α or β/α/α units.
The TIM barrel fold is a well-known domain formed by repeating β/α motifs.
Classification with CATH
Protein domains grouped into classes based on secondary structure composition (mostly α, mostly β, mixed α+β, little α/β).
Further organized by architecture, topology (connection order), and homologous superfamilies (similar evolutionary origin).
Example: "Rossman fold" for NAD(P)-binding enzymes.
General Principles of Globular Proteins
Have defined hydrophobic core and hydrophilic surface.
β sheets often twisted or form barrels.
Polypeptide chains turn with β turns and γ turns, important for compact folding.
Proline frequently found in turns or breaks helices.
Irregular and Unstructured Regions
Not all parts fit typical secondary structures; loops and irregular folds occur.
Some proteins have intrinsically unstructured regions important for signaling and interaction versatility.
Primary Sequence Determines Folding
The amino acid sequence encodes the information needed for a protein to fold into its unique three-dimensional structure.
Classic experiments (e.g., by Anfinsen with RNase A) show a denatured protein can refold spontaneously to its native structure when returned to physiological conditions, indicating sequence alone is sufficient to guide folding
3 Forces Stabilizing Secondary and Tertiary Structures
Conformational Entropy
Enthalpic Interactions (ΔH)
Hydrophobic Effects (Favorable Entropy)
Conformational Entropy
Folding reduces the protein's conformational entropy (randomness), which disfavors folding (ΔS < 0).
This entropy loss is a barrier to folding that must be overcome by favorable interactions
Enthalpic Interactions consist of which 2 bonding types
Hydrogen Bonding
Van Der Waals Interactions
Enthalpic Interactions (ΔH)
Favorable noncovalent interactions within the folded protein contribute negative ΔH (stabilizing).
These include:
Hydrogen bonds (both backbone and side chain).
Ionic/salt bridges (charge–charge interactions between oppositely charged side chains).
Van der Waals interactions (weak dipole-induced dipole attractions).
Some proteins also form disulfide bonds (covalent S–S bonds between cysteines), greatly stabilizing the structure post folding.
Hydrophobic Effect (Favorable Entropy)
Hydrophobic side chains cluster inside the protein away from solvent water.
This burial of hydrophobic groups releases structured water molecules into the bulk solvent, increasing solvent entropy (ΔS > 0), which favors folding.
The hydrophobic effect is a dominant driving force for folding in water.
Overall Thermodynamic Balance
The net free energy change for folding (ΔG = ΔH – TΔS) is negative due to a combination of:
Loss of conformational entropy (unfavorable).
Gain in enthalpy from internal bonding (favorable).
Gain in entropy of the solvent due to hydrophobic effect (favorable).
This delicate balance explains why proteins fold spontaneously under physiological conditions but are vulnerable to denaturation by heat, pH changes, or chemical agents.
Summary Table: Forces Affecting Protein Folding
Factor | Effect on Folding | Why/How |
|---|---|---|
Conformational entropy | Opposes folding (ΔS < 0) | Folding reduces flexibility and randomness |
Noncovalent bonding | Favors folding (ΔH < 0) | Hydrogen bonds, ionic interactions, van der Waals forces |
Hydrophobic effect | Favors folding (ΔS > 0) | Burial of hydrophobic side chains frees structured water |
Disulfide bonds | Favors stability post folding | Covalent bonds strengthen folded protein |
Globular Protein Folding proceeds through…
intermediate states such as the molten globule, which has native-like secondary structure but lacks full tertiary interactions.
The energy landscape (folding funnel) model explains…
the folding trajectory as a thermodynamically downhill path narrowing from many unfolded conformations to fewer folded ones.
Globular Folding involves…
nucleation of secondary structures (like α helices) and hydrophobic collapse, where hydrophobic residues cluster internally to avoid water.
Chaperones in cells
assist folding by providing isolated environments to prevent aggregation and help proteins fold correctly. These chaperones use ATP to mediate the folding cycle
Misfolded proteins can aggregate into…
amyloid fibrils, linked with diseases like Alzheimer’s, Parkinson’s, and prion diseases.
The prion protein (PrP)…
can propagate disease by inducing conformational change in normal protein to a β-sheet rich pathogenic form.
What is the current state of protein secondary and tertiary structure prediction?
Secondary structure prediction (α helix, β sheet, turns) is about 70–85% accurate using empirical and computational methods based on known structures and amino acid propensities.
Tertiary structure prediction is more challenging due to long-range interactions but recent AI-based tools (e.g., AlphaFold, RoseTTAFold, trRosettaX) have made significant progress with near atomic-level accuracy for some proteins.
Reliable prediction of complete 3D structure from sequence alone remains an active area of research but is rapidly improving with deep learning approaches.
Quaternary Structure of Proteins
Quaternary structure is the assembly of two or more folded polypeptide chains (subunits) into a specific functional protein complex.
2 types of way quaternary structure of proteins can be formed
Homotypic interactions
Heterotypic interactions
Homotypic interactions:
Association of identical or nearly identical subunits (e.g., hemoglobin with four identical subunits).
Heterotypic interactions:
Association of different subunits with distinct structures (e.g., GroEL and GroES chaperonin complex).
The types of molecular interactions stabilizing quaternary structure are similar to tertiary structure (5):
Salt bridges (electrostatic interactions)
Hydrogen bonds
Van der Waals forces
Disulfide bonds (sometimes)
Hydrophobic effects
Primary Protein Structure Bonds/Interactions Involved
Peptide bonds (covalent bonds linking amino acids in a chain)
Secondary Protein Structure Bonds/Interactions Involved
Hydrogen bonds between backbone amide N–H and carbonyl C=O
Tertiary Protein Structure Bonds/Interactions Involved
Hydrogen bonds (backbone & side chains)
Ionic Bonds (salt bridges between charged side chains)
Van Der Waals Interactions (close packing of nonpolar side chains)
Disulfide Bonds (covalent S–S bonds between cysteine residues)
Hydrophobic Interactions (burial of nonpolar side chains inside)
7 Spectroscopic Methods for Protein Structure
UV-Visible Absorption Spectroscopy:
Fluorescence Spectroscopy:
Circular Dichroism (CD) Spectroscopy:
Infrared (IR) and Raman Spectroscopy:
Nuclear Magnetic Resonance (NMR) Spectroscopy:
X-ray Crystallography
Mass Spectrometry (MS):
UV-Visible Absorption Spectroscopy:
Used to measure protein concentration via absorption at 280 nm (aromatic amino acids) and monitor certain prosthetic groups (heme, metals).
Changes in absorption can indicate conformational or environmental changes.
Fluorescence Spectroscopy:
Tryptophan and tyrosine residues fluoresce; fluorescence changes indicate folding/unfolding or environment polarity shifts.
Used in live cell imaging with fluorescent tags like GFP
Circular Dichroism (CD) Spectroscopy:
Sensitive to secondary structure (α helix, β sheet, random coil) by differential absorption of circularly polarized light.
Can monitor protein folding/unfolding in solution.
Infrared (IR) and Raman Spectroscopy:
Provide vibrational information about backbone and side chains, useful for secondary structure analysis.
Nuclear Magnetic Resonance (NMR) Spectroscopy:
Provides atomic-level information about protein structure and dynamics in solution.
Detects chemical environments of nuclei; best for smaller proteins (<50 kDa).
Produces ensembles of structures reflecting conformational flexibility.
X-ray Crystallography:
Gold standard for high-resolution 3D structure determination of crystallized proteins.
Difficult for flexible or unstructured proteins that don’t crystallize well.
Mass Spectrometry (MS):
Used for primary structure (sequence identification) and higher-order structural information like protein-protein interactions, folding states, and complex assemblies.
Often combined with other methods (cryo-EM) to study large complexes.
4 Methods to Determine Protein Molecular Mass and Subunit Composition
Size Exclusion Chromatography (SEC):
Analytical Ultracentrifugation (Sedimentation Equilibrium)
Analytical Ultracentrifugation (Sedimentation Equilibrium):
SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE):
Size Exclusion Chromatography (SEC):
Separates proteins by size (hydrodynamic radius).
Estimates native molecular mass in near-physiological conditions.
Shape of protein can affect accuracy.
Analytical Ultracentrifugation (Sedimentation Equilibrium):
Measures sedimentation-diffusion equilibrium to determine molecular mass accurately under native conditions.
Mass Spectrometry (MS):
Highly accurate molecular mass determination of protein subunits.
Can be used after isolation of native complexes by SEC
SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE):
Proteins are denatured and coated with negatively charged SDS molecules.
Electrophoretic mobility depends on length (size) only.
Runs under reducing (with β-mercaptoethanol) and non-reducing conditions distinguish disulfide-linked subunits.
Comparison with known MW markers allows estimation of subunit sizes.
Multiple bands indicate multiple types of subunits; single bands suggest identical or single chains.