4: mechanistic organic chem
1/157
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
158 Terms
what is the relationship between s/p character and reactivity?
higher s character = electrons held closer to the nucleus = less reactive
higher p character = electrons held further from the nucleus = more reactive
draw the molecular orbital diagram from a carbonanion
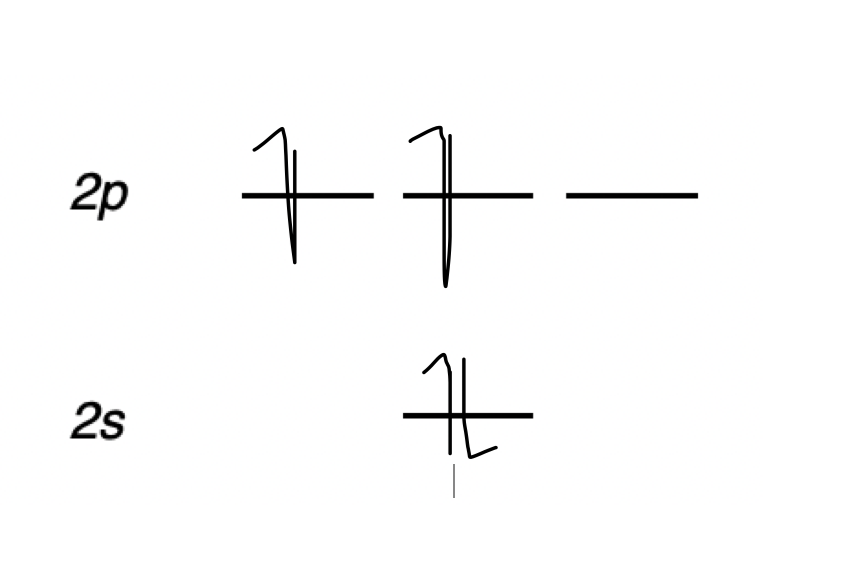
describe the different hybridisations of carbon and their relative reactivities
the extent of p orbital contribution depends on the amount available i.e. alkynes have 2 p orbitals involved in triple bond.
2s + 2p(x) = 2 sp hybrid orbitals = linear
2s + 2 2p = 3 sp2 hybrid orbitals = trigonal planar
2s + 3 2p = 4 sp3 hybrid orbitals = tetrahedral
sp = 50% s 50% p = ~unreactive
sp2 = 33% s 66% p
sp3 = 25% s 75% p = ~reactive
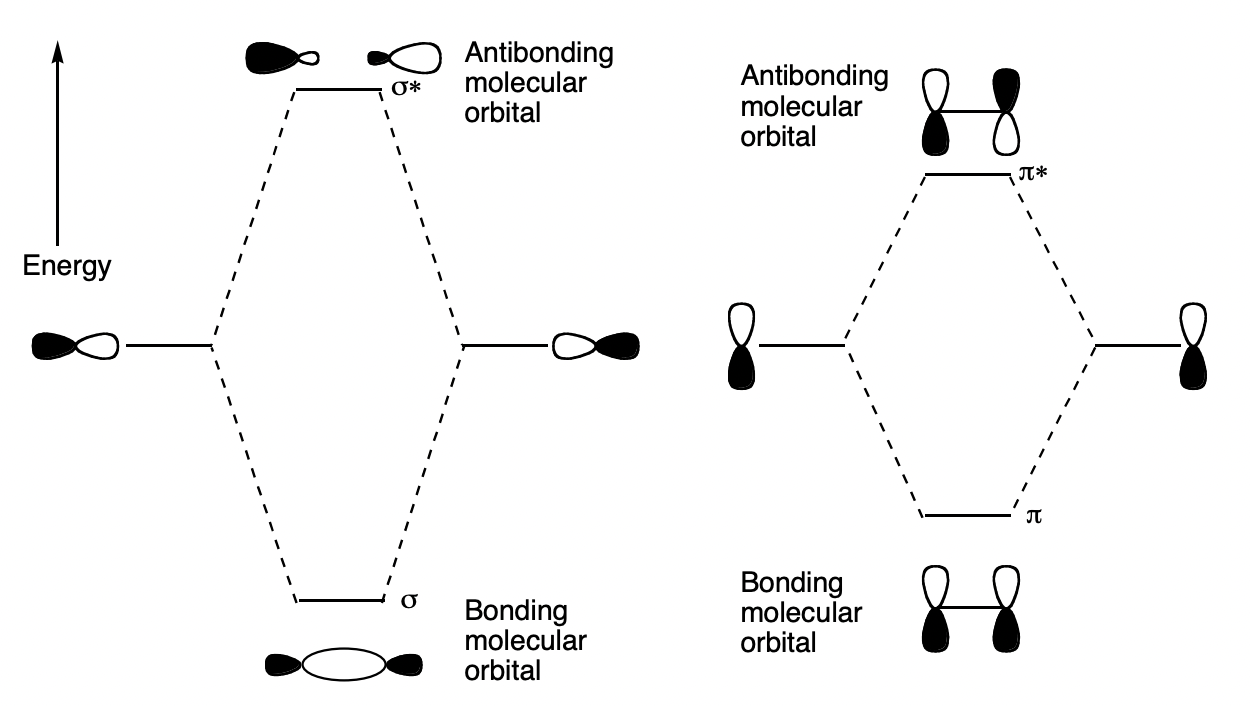
draw the arrangement of orbitals that gives rise to ∏ and sigma bonding
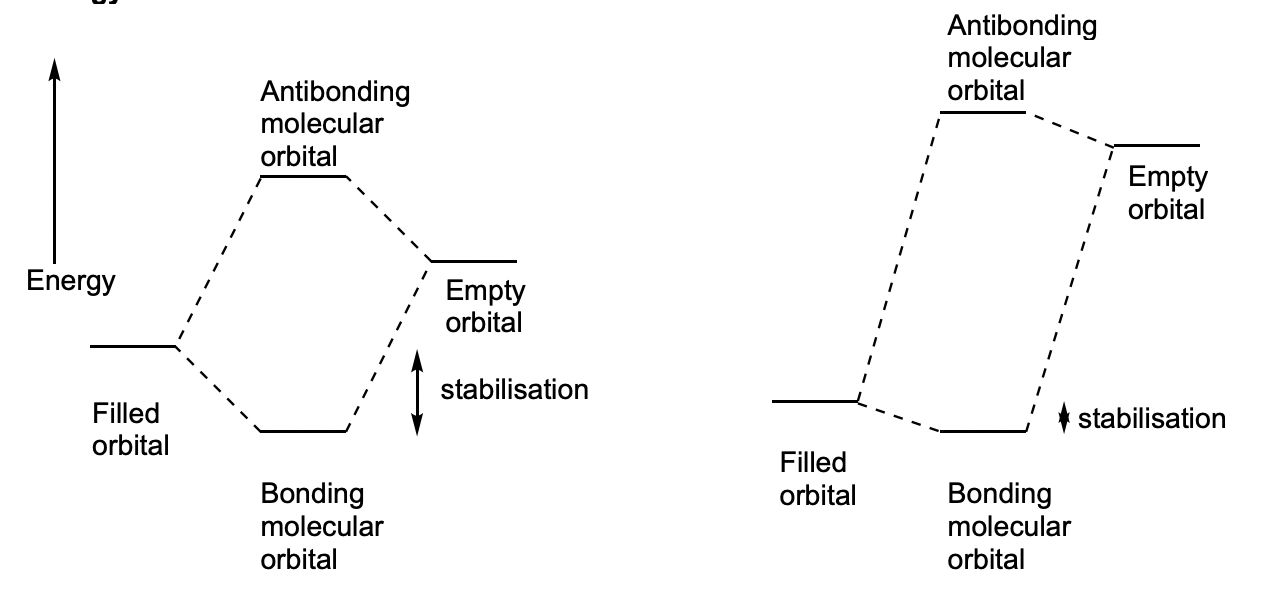
how does the separation of filled and empty orbitals affect stabilisation?
small separation = good stabilisation
large seaparation = poor stabilisation
what are the (main) conditions for orbital interaction?
matched symmetry
good overlap
similar energies
describe lewis acids and lewis bases in terms of orbitals
lewis base = donate electron density = high energy (unstable) HOMO
lewis acid = accept electron density = low energy (stable) LUMO
explain why lewis adducts are so stable
lewis base = high energy HOMO
lewis acid = low energy LUMO
in conjunction, this minimise the minimise the orbital separation and increase stabilisation
what is Bredit’s rule? explain
a double bond is not possible at the bridgehead of two small rings
= poot orbital overlap
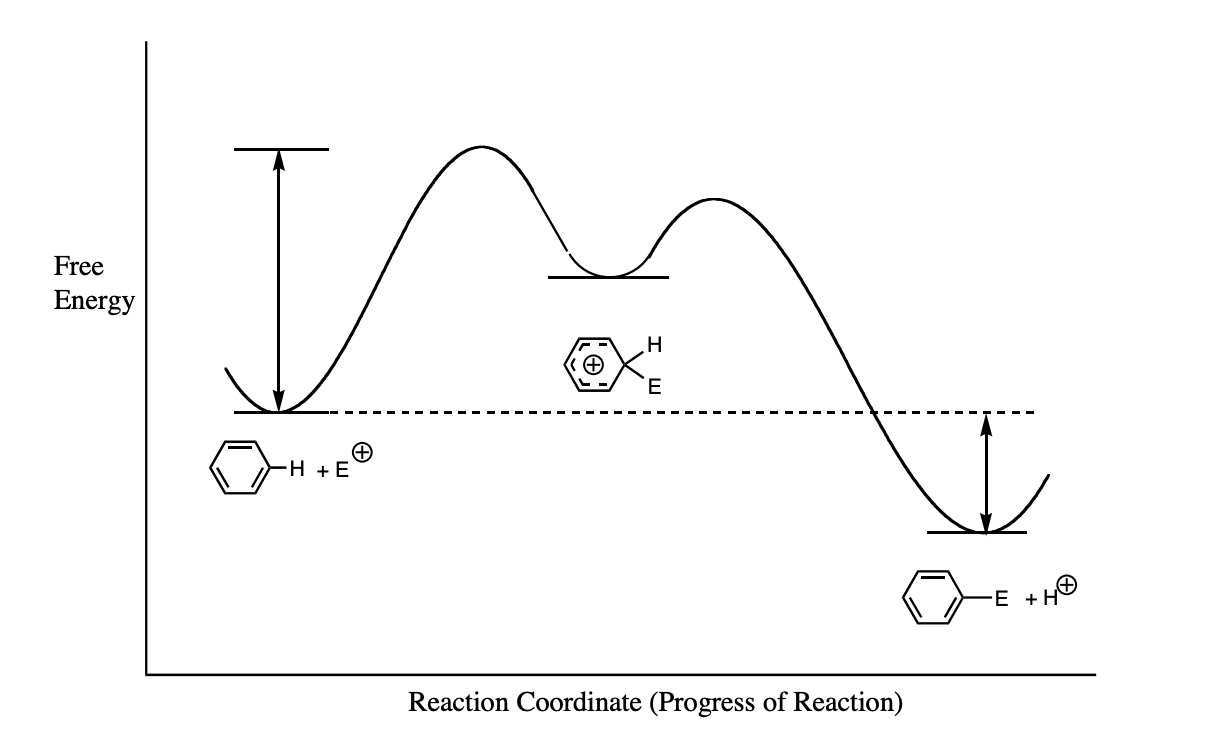
label this reaction coordinate diagram
define a transition state
= maxima on the reaction coordinate diagram
= partially formed bonds
= does not exist for finite time and cannot be isolated/observed
define an intermediate
= exists between energy barriers in “well” on reaction coordinate diegram
= fully formed bonds
= isolatable/observable
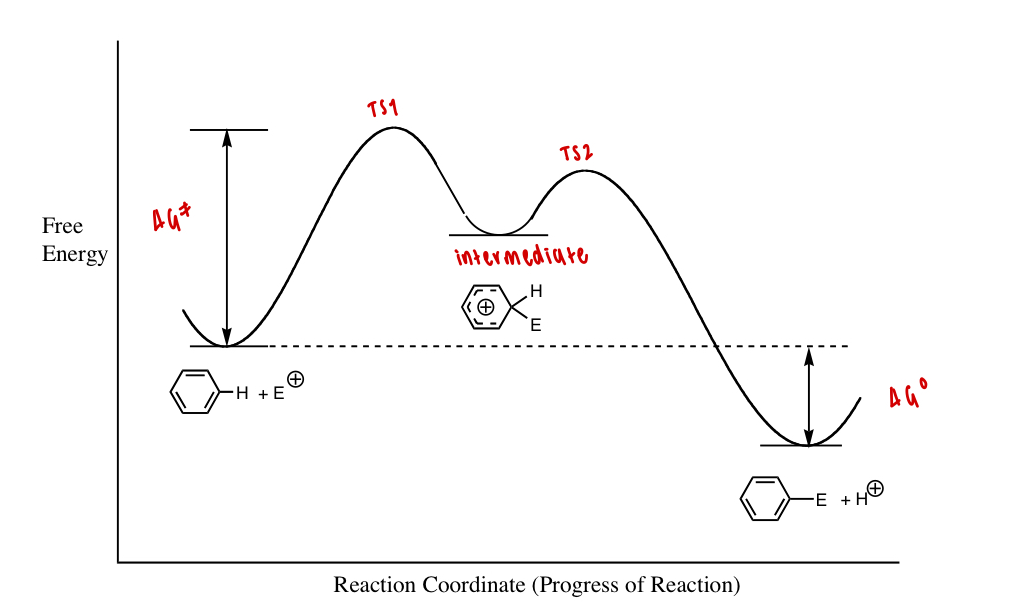
define the ΔG° for a reaction in terms of entropy/enthalpy

define the ΔG° for a reaction at equilibrium
defines the position of equilibrium
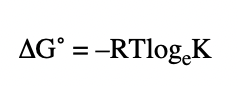
how does ΔG° control the spontaneity of reaction
-ve ΔG° favours the forward reaction and product formation
+ve ΔG° favours the backwards reaction and reactant formation
how can ΔG° be altered?
-ve ΔH°:
stronger bonds forming (-ve H) than breaking (+ve H)
less strain in products than reactant
+ve ΔS°
increase in disorder is favoured
describe how choice of polar solvent can favour products or reactants
polarity: reactants > products
polar solvent favours reactants
polarity: products > reactants
polar solvent favours products
polar solvents cluster around charges in reactants and products (solvation) so that charge is delocalised.
solvation = interactions formed = -ve H = favoured
what are the two types of solvent?
protic
= contain O-H or N-H
= solvate +ve and -ve charges
aprotic
= do not contain O-H or N-H
= solvate only +ve charges
describe the dielectric constant
= measure of polarity
large dielectric constant = polar
small dielectric constant = non-polar
= measure of ability to solvate charge
examples of protic solvents
h2o, meoh, etoh, tbuoh, acoh
examples of aprotic solvents
dmso, dmf, acn or mecn, dcm, thf, etoac, et2o
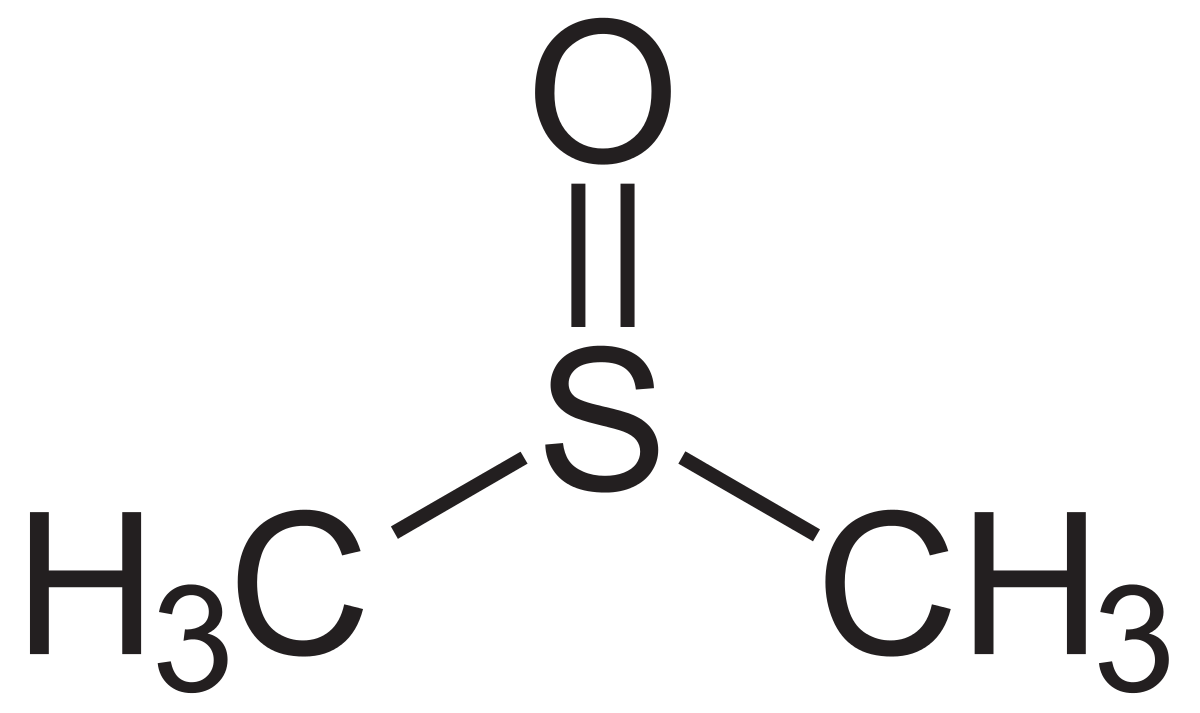
draw the structure of DMSO
draw the structure of DMF
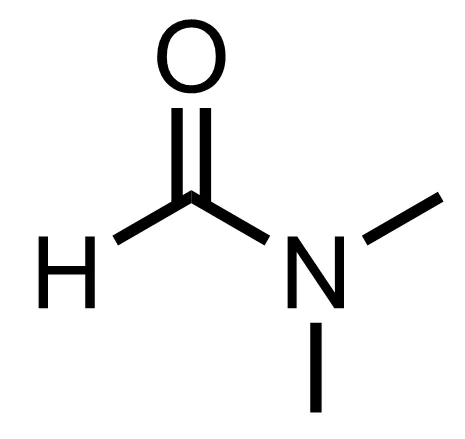
draw the structure of ACN/MeCN
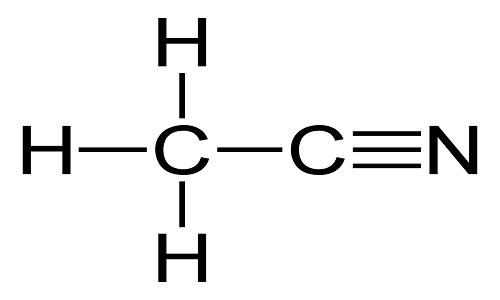
draw the structure of THF
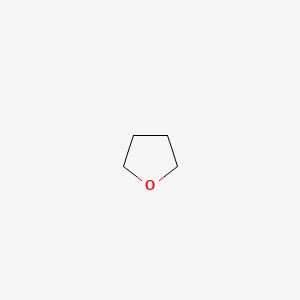
rank protic solvents in terms of solvation ability
H2O > MeOH > EtOH > tBuOH > AcOH (delocalised = weaker)
rank aprotic solvents in terms of solvation ability
DMSO > DMF > ACN/MeCN > acetone > DCM > THF> EtOAc > Et2O
what are the 4 types of solvent; describe each
polar protic = +ve & -ve = good solvation
polar aprotic = +ve = good solvation
apolar protic = +ve & -ve = poor solvation
apolar aprotic = +ve = poor solvation
what is the rate determining step?
= step with largest activation energy
= determines the overall rate of reaction
what does the rate of the RDS depend on?
number of collision between reacting molecules in a given period
fraction of collisions with sufficient energy for reaction
fraction of collisions with correct orientation for reaction
describe the role of the RDS is determining the order of reaction
reactants involved prior to and in the RDS influence the rate of reaction.
reactants involved after the RDS will not influence the rate of reaction.
describe the rate constant
fundamental property of a reaction
depends on:
temperature
pressure
solvent
DOES NOT DEPEND ON CONCENTRATION
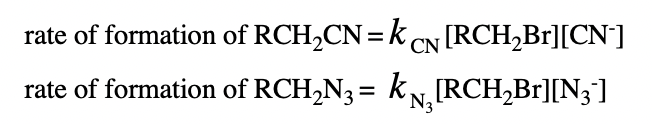
give an equation for the ratio of product for the concurrent reactions described by the equations
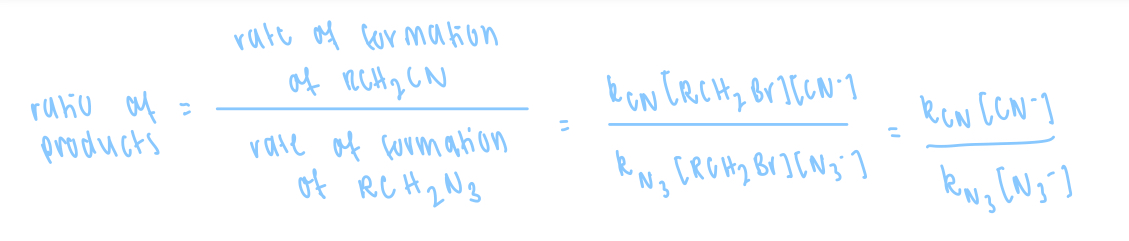
![<p>define the rate of formation when:</p><ul><li><p>[CN-] = [N3-]</p></li><li><p>32 x [CN-] = [N3-] </p></li></ul><p><strong>given k[CN] = 16 k[N3]</strong></p>](https://knowt-user-attachments.s3.amazonaws.com/d2ea90b6-7c0c-4885-8467-535f2144c746.png)
define the rate of formation when:
[CN-] = [N3-]
32 x [CN-] = [N3-]
given k[CN] = 16 k[N3]

define molar free energy of activation, ΔG‡
= difference in energy between a ground state and a transition state
= related to the rate constant, k.
~ analogous to activation energy
how do ΔG‡ and E(a) differ?
ΔG‡ = contains ΔS‡ and ΔH‡
E(a) = contains ΔH‡
give an equation that relates rate constant with ΔG‡
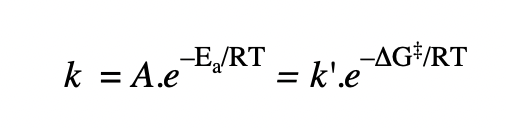
ΔG‡ = ΔS‡ and ΔH‡ related
vs
Ea = ΔH‡ related
A = ΔS‡ related
small |ΔG‡| = large k = fast reaction
large |ΔG‡| = small k = slow reaction
= analogous to activation energy
what are the ranges for enthalpy and entropy of activation?
ΔH‡ = 40 - 125 kJ mol(-1)
ΔS‡ = -150 - 60 J K(-1) mol(-1)
ΔH‡ = always positive due to breaking of bonds without reforming (forming TS) = always disfavoured
smaller = ~favoured
larger = ~disfavoured
ΔS‡:
-ve = disfavoured
+ve = favoured
describe a dissociate RDS in terms of enthalpy and entropy
favoured by entropy = increase in disorder
disfavoured by enthalpy = bonds broken
describe an associative RDS in terms of enthalpy and entropy
disfavoured by entropy = increase in order
favoured by enthalpy = balance in bond breaking/forming
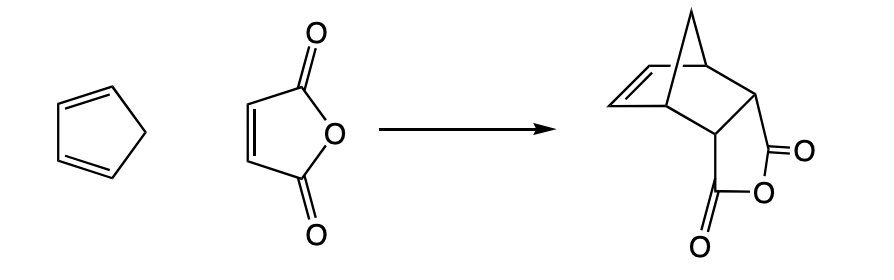
describe.a diels alder reaction in terms of entropy
requires simultaneous bond formation = highly ordered TS
associated RDS
highly disfavoured by entropy
define an elementary reaction
= a single step in a reaction mechanism proceeding in a particular direction
define molecularity of an elementary step
= the number of molecules involved in the formation of a single transition state (ignoring those only involved in solvation)
= may be different depending on direction
what are the two molecularities of elementary steps?
unimolecular
bimolecular
molecularities above this are exceedingly slow since they require the simultaneous collision of ≥3 molecules
describe microscopic reversibility
= the mechanism of the (lowest energy) reverse reaction must retrace each step of the mechanism of the (lowest energy) forward reaction (in microscopic detail)
= RDS are mirrored i.e. formation of intermediate in both directions
how can reactions be accelerated?
increase concentration(s) of reactant(s)
increase temperature
reduce ΔG‡ of the RDS = raise ground state energy and/or lower TS energy
describe SN2 reactions
bimolecular; second order
concerted; 1 step
inversion of configuration
draw the mechanism of an SN2 reaction
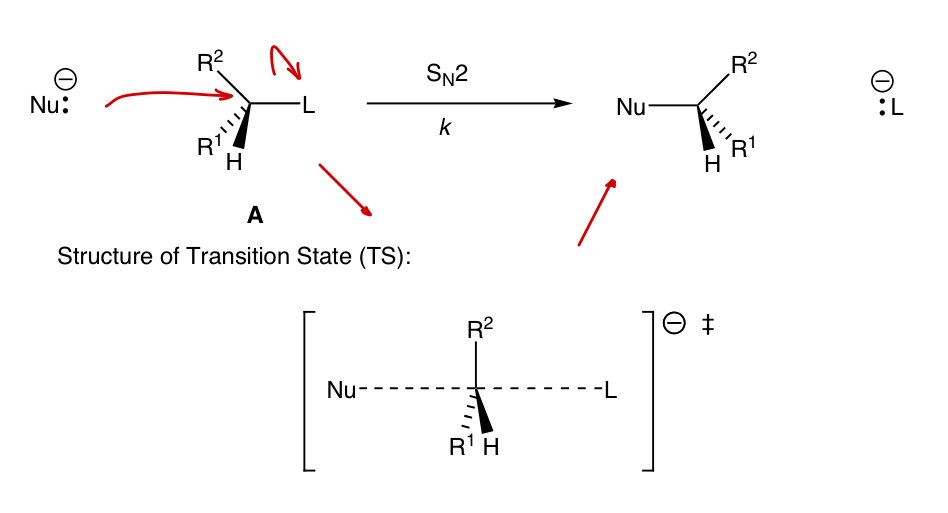
describe SN2 reactions in terms of ΔS‡ and ΔH‡
associative TS
disfavoured by ΔS‡
favoured by ΔH‡
draw a reaction coordinate diagram of an SN2 reaction
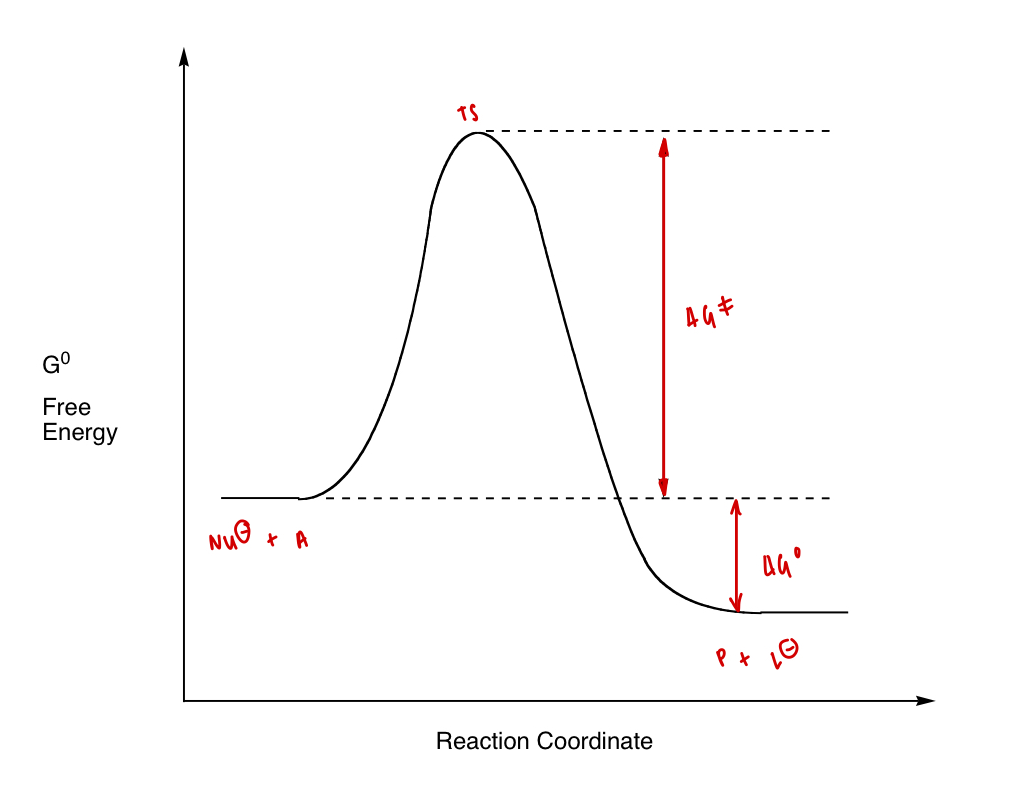
what can be determined from ΔG‡ vs ΔG°?
ΔG‡ = rate constant, k = RDS varies rate
ΔG° = equilibrium constant, K = varies position of equilibrium
draw the MO diagram for the transition (Nu-C)
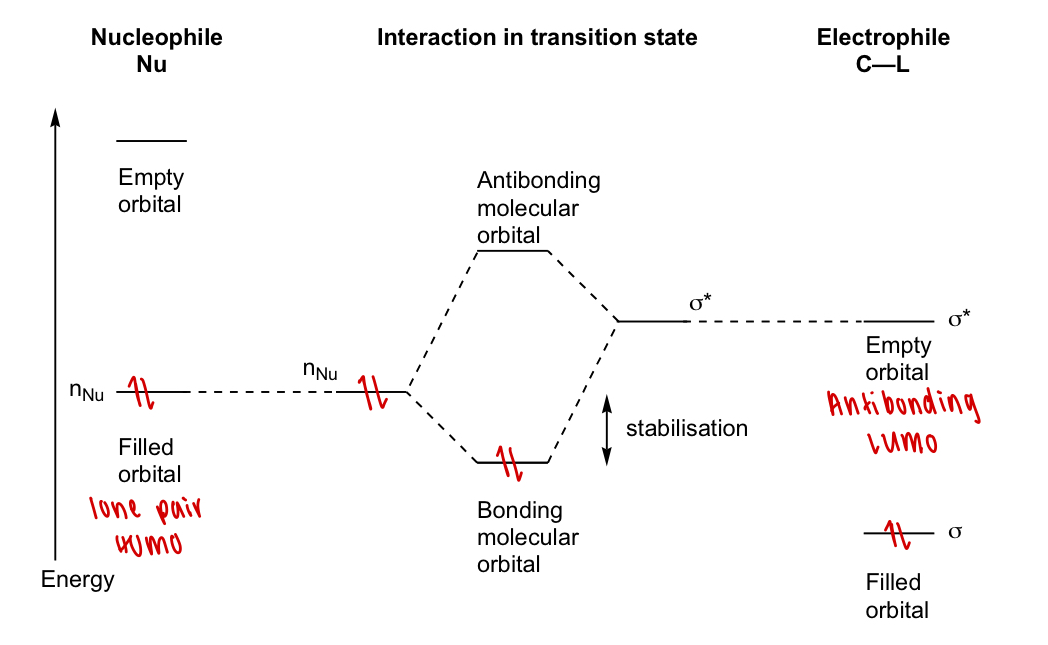
describe SN2 reaction in terms of orbital overlap
back lobe (opposite to L):
phase matched
good overlap
front lobe (at L):
phases matched
poor overlap
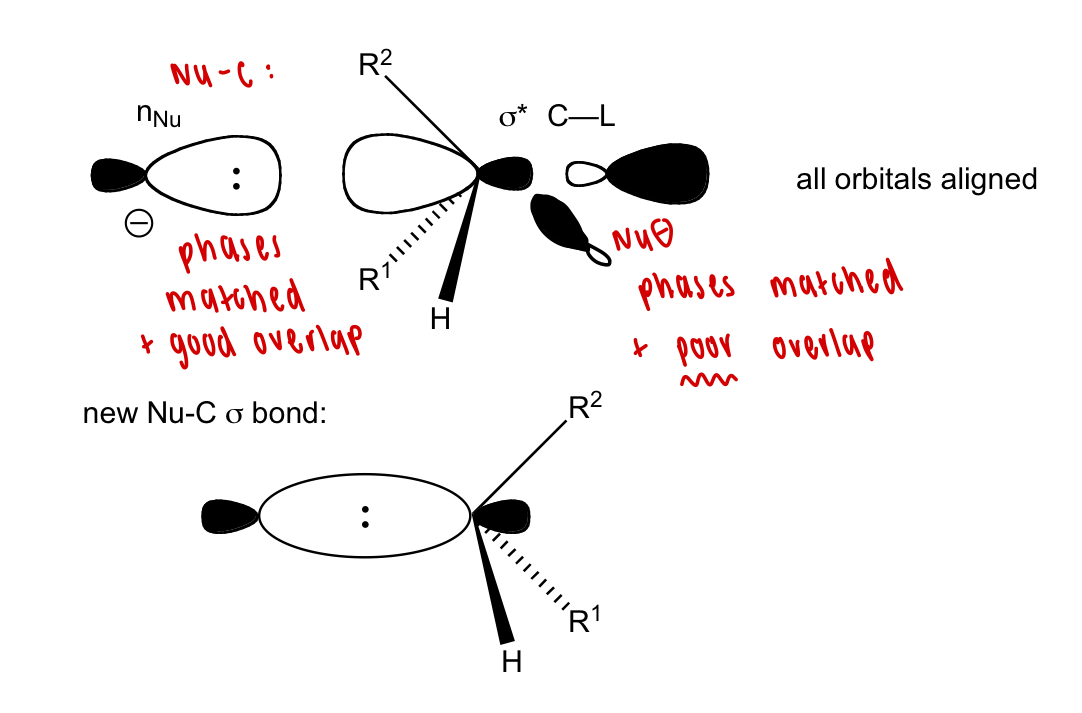
why is Nu attack favoured from the back lobe/inhibited at the front lobe?
poor orbital overlap/alignment
electrostatic repulsions from L and R
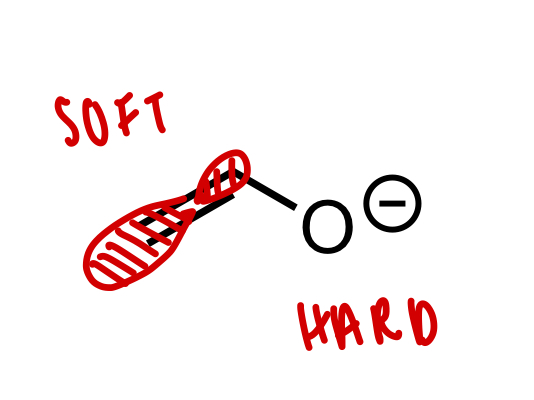
describe soft and hard electrophiles/nucleophiles
soft = polarisable = large, uncharged, diffuse
hard = less polarisable = small, highly charged
examples of soft and hard electrophiles/nucleophiles
nucleophiles
soft = I(-), RS(-), NC(-) = large attacking atom/charge distribution over atoms
hard = F(-), RO(-) = small attacking atom/concentrated charge
electrophiles
soft = alkyl halide (large halide i.e. I)
hard = H(+), alkyl halide (small halide i.e. Cl), carbonyls, SiMe3(+)
describe the energy of soft/hard electrophile LUMO/nucleophile HOMO
electrophiles:
soft = low LUMO
hard = high LUMO
nucleophiles:
soft = high HOMO
hard = low HOMO
what kind of electrophiles are alkyl halides and what kind of NUCLEOPHILE IS FAVOURED IN SN2?
alkyl halides = soft electrophiles = low LUMO
interact best with soft nucleophiles = high HOMO
i.e. I(-), RS(-), NC(-)
describe enolates variable nucleophilicity
soft at B carbon
hard at charged O
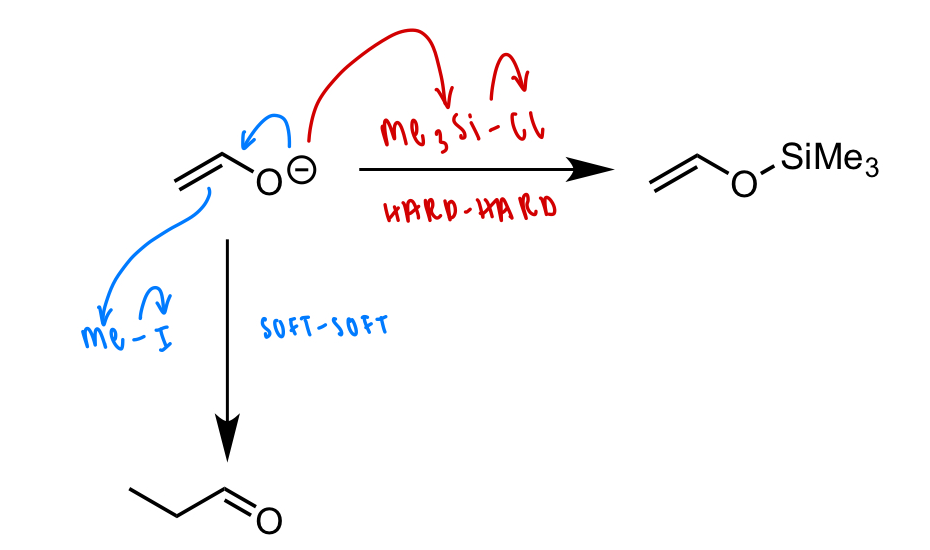
draw the reaction of enolate with:
silylating agents (Me3Si-Cl)
alkyl halides (Me-I)
describe the speed of SN2 reactions on different °s of alkyl halides
1° = YES; fast
2° = majorly no; if yes, slow
3° = NO
described the speed of SN2 reactions on different length 1° alkyl halides
smaller chain = faster SN2
describe the speed of SN2 reactions with different size of nucleophiles
bulky nucleophiles = slow
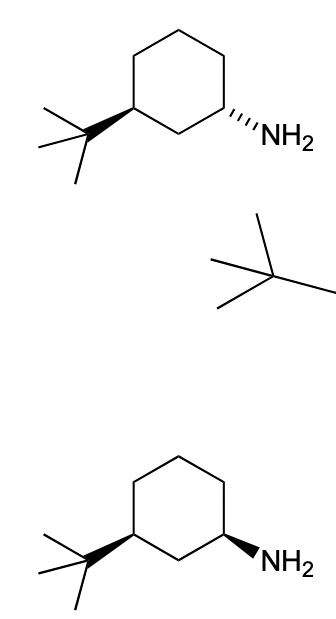
describe the SN2 reaction of Me-I with these nucleophiles
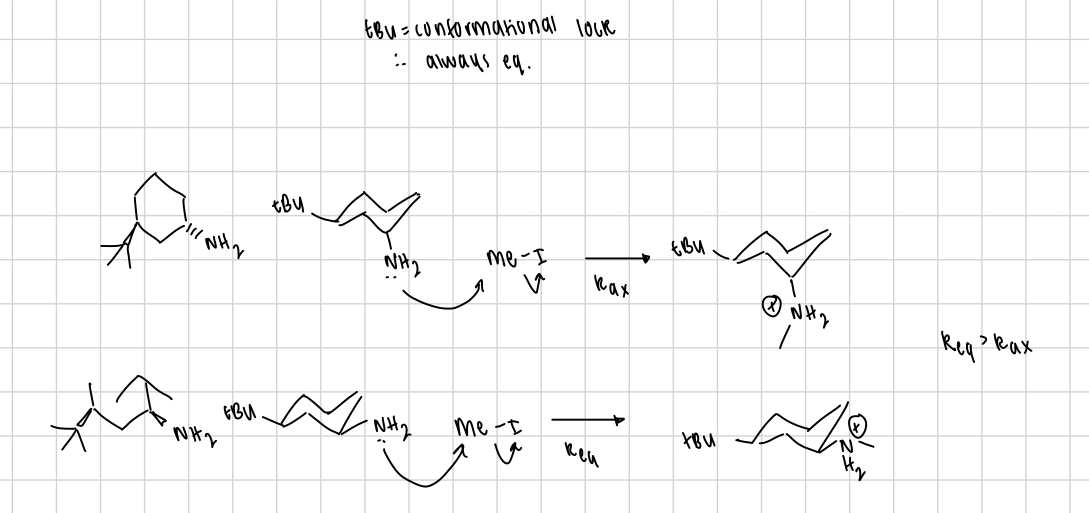
what can reduce ΔG‡ w.r.t. leaving group
weak C-L bond
L(-) stability
what acts as a proxy for leaving group ability?
pKa
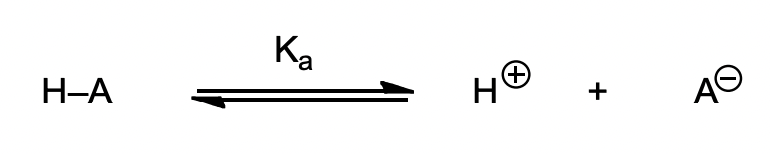
= defines the stability of the anion
weak acids: tend to protonated form
strong H-A bond
poor stabilisation of A(-)
strong acids: tend to deprotonated form
weak H-A bond
good stabilisation of A(-)
therefore:
strong acid = small pKa = good leaving groups
weak acid = large pKa = poor leaving groups
define pKa and how it relates to acid strength
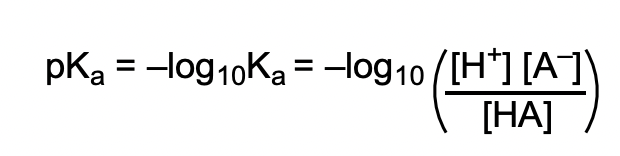
pKa ∝ 1/acid strength
describe the relative leaving group abilities of the halides
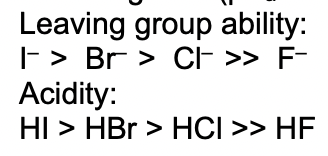
give examples of poor leaving groups
F(-)
RS
CN
OH
OR
describe why there is no SN2 at sp2 (majorly) or sp (ever) atoms
line of attack is blocked
C has higher s character = stronger C-X bond
C-X sigma bond often not the LUMO
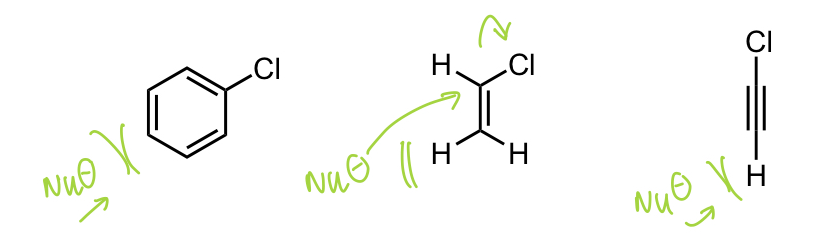
what can reduce ΔG‡ w.r.t nucleophile
strong C-Nu bond
Nu instability/reactivity
what acts as a proxy for nucleophilicity (SAME ATOM/SAME SIZE)?
pKa of conjugate acid
weak conjugate acids: tend to protonated form
= conjugate base is less stabilised (less likely to be protonated) by weak conjugate acid
strong acids: tend to deprotonated form
= conjugate base is more stabilised (likely to be protonated) by strong conjugate acid
therefore:
strong conjugate acid = small pKa = poor nucleophile
weak conjugate acid = large pKa = good nucleophile
examples of nucleophiles and relative reactivities
most nucleophilic → least nucleophilic
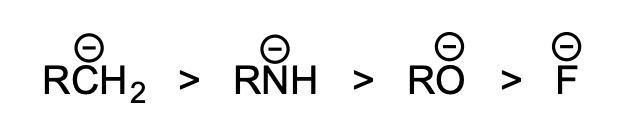
summarise pKa and LG/Nu ability
acid strength ∝ 1/pKa ∝ LG ability
conjugate acid strength ∝ 1/pKa ∝ 1/Nu ability
describe the variable solvation of charges by protic solvents
hard negative charges = strong charge-dipole interactions = good solvation
soft negative charges = weak charge-dipole interactions = poor solvation
what does the nucleophilicity of attacking atoms of different sizes depend on?
solvent
aprotic solvents:
negative charges are not solvated = nucleophilicity ∝ basicity
protic solvents:
hard negative charges are solvated = weakly nucleophilic
(charge is delocalised in TS so less interaction with solvent)
soft negative charges are poorly solvated = strongly nucleophilic
for what kind of SN2 reactions are polar aprotic solvents particularly good for?
uncharged electrophile + charged nucleophile
polar aprotic solvents only solvate +ve charges. +ve component/counterion of nucleophile is well solvated while the -ve part is poorly solvated = naked and highly reactive.
what kind of solvent is preferred in:
charged nucleophile + uncharged electrophile
uncharged nucleophile + uncharged electrophile
charged nucleophile + uncharged electrophile
= (polar) aprotic
= TS has delocalised charge w.r.t reactants/products
uncharged nucleophile + uncharged electrophile
= polar (aprotic or protic)
= TS/products is more polar than reactants = stabilised by polar solvent
what is used to facilitate SN2 between non-polar RX and ionic compounds?
2 immiscible solvents
phase transfer catalyst
example of a phase transfer catalyst
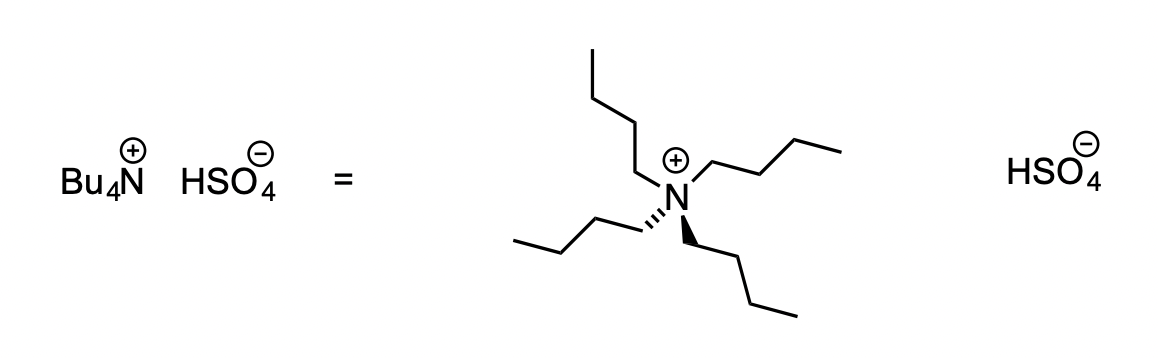
= soluble in both organic and aqueous phases
describe phase transfer catalysis
non-polar RX + ionic compound + 2 immiscible solvents
non-polar RX will dissolve in organic phase; ionic compound will dissolve and ionise in aqueous phase
phase transfer catalyst drags -ve ion into organic phase to balance charge; -ve ion is naked and highly reactive = SN2
phase transfer catalyst coordinates to -ve ion product of SN2 and drags back into aqueous phase
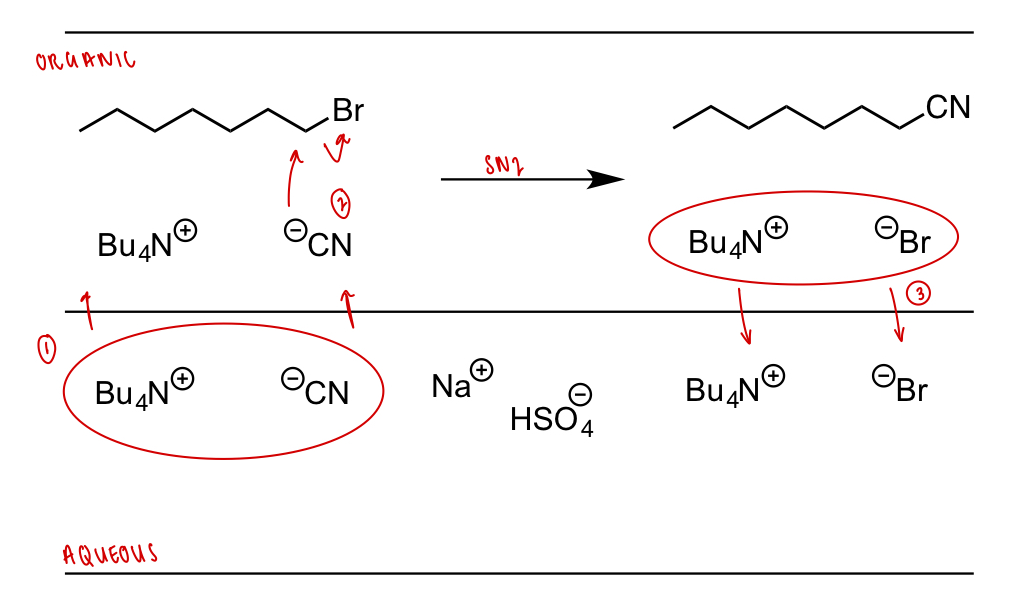
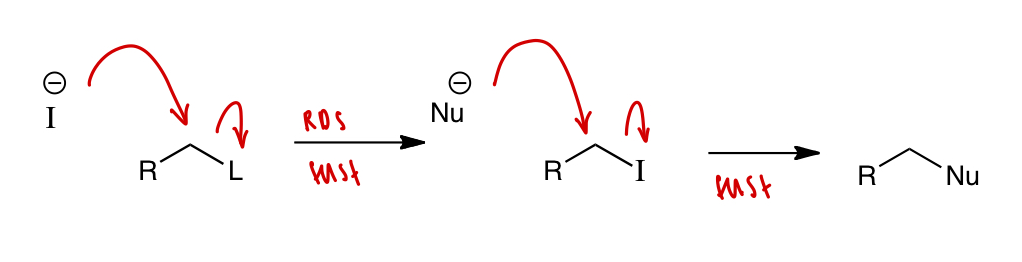
what can be used as a nucleophilic catalyst and explain
iodide I(-)
nucleophilicity in protic solvent: I(-) > Nu(-)
I(-) = large anion = soft = poorly solvated by protic solvent
iodide displaces L faster than Nu(-)
leaving group ability: I(-) > L
HI = strong acid = good LG
Nu(-) displaces in R-I faster than in R-L
why are cyclisations favoured over bimolecular reactions?
by entropy
cyclisations:
dissociative TS
ΔS = 0
bimolecular:
associative TS
ΔS = -ve (loss of translational entropy via associative TS)
relative reactions rate for different ring sizes (cyclisations)
5 > 6 > 3 > 4 & 7 > other rings
why do cyclisation forming small (3/4) rings have slow reaction rates?
ring strain:
in products increases ΔH*
in TS increases ΔH‡
why do cyclisation forming large (>6) rings have slow reaction rates?
probability of ends colliding with correct orientations is small
what is effective molarity
EM = k(intra) / k(inter)
unimolecular: the concentration of [A] required to match k(intra)
why would effective molarity (EM) be low?
favourable orbital alignment/other stereoelectronic factor present in inter absent in intra
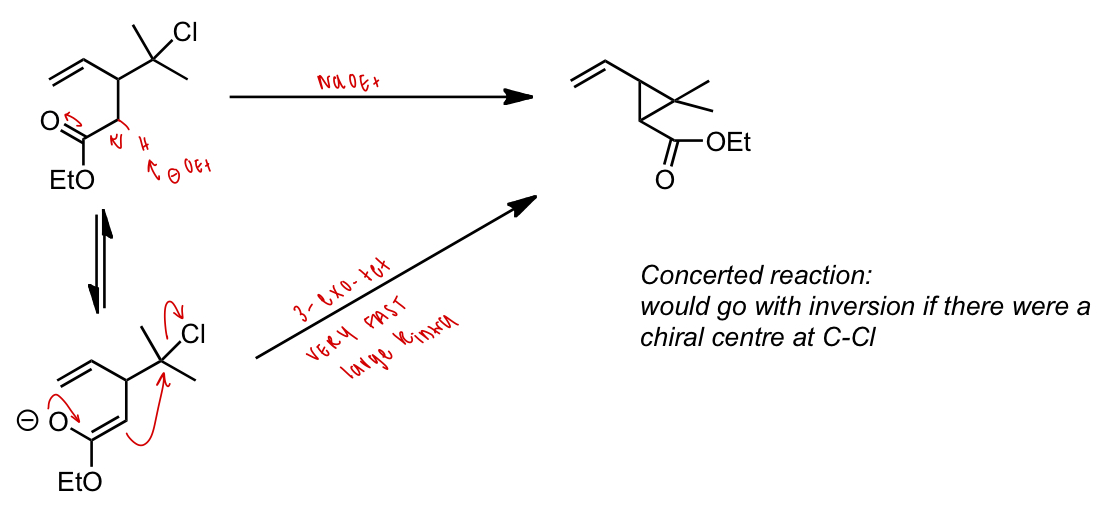
describe how reaction would proceed here with NaOEt
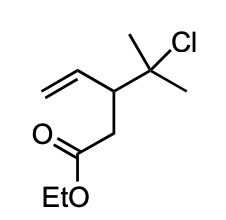
SN2 at 3° C-X possible due to favoured 3 membered ring
describe intra SN2 on cyclohexane
Nu and LG must be 1,2-trans diaxial and antiperiplanar
= orbital alignment
what does X-exo/endo-trig/tet mean?
X = ring size in the TS
exo/endo:
exo = bond broken outside ring of TS
endo = bond broken within ring of TS
trig/tet:
trig = LG attached to trigonal C (sp2)
tet = LG attacked to tetrahedral C (sp3)
what are Baldwin’s guidelines to favourable cyclic reactions
exo > endo
= favoured by good orbital alignment
3 to 7-exo-tet > 3 to 7-endo-tet
what form does the lowest energy TS of an SN2 take?
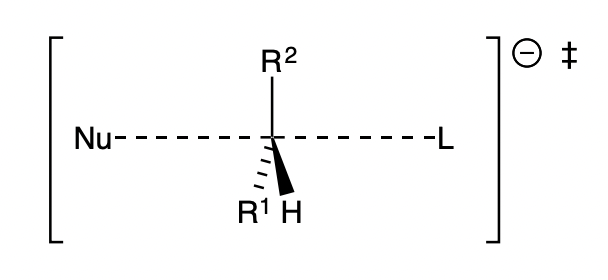
= linear
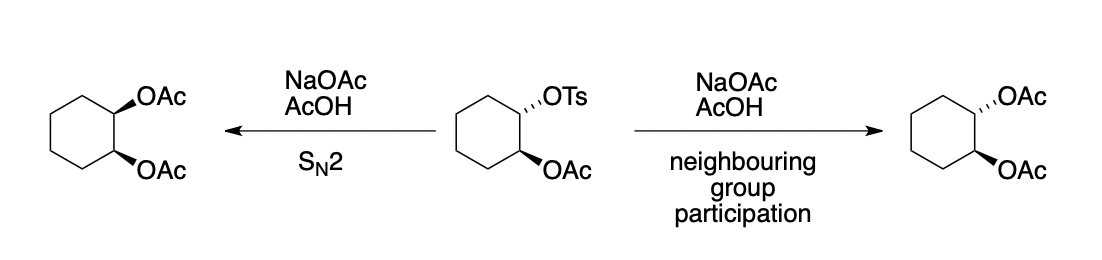
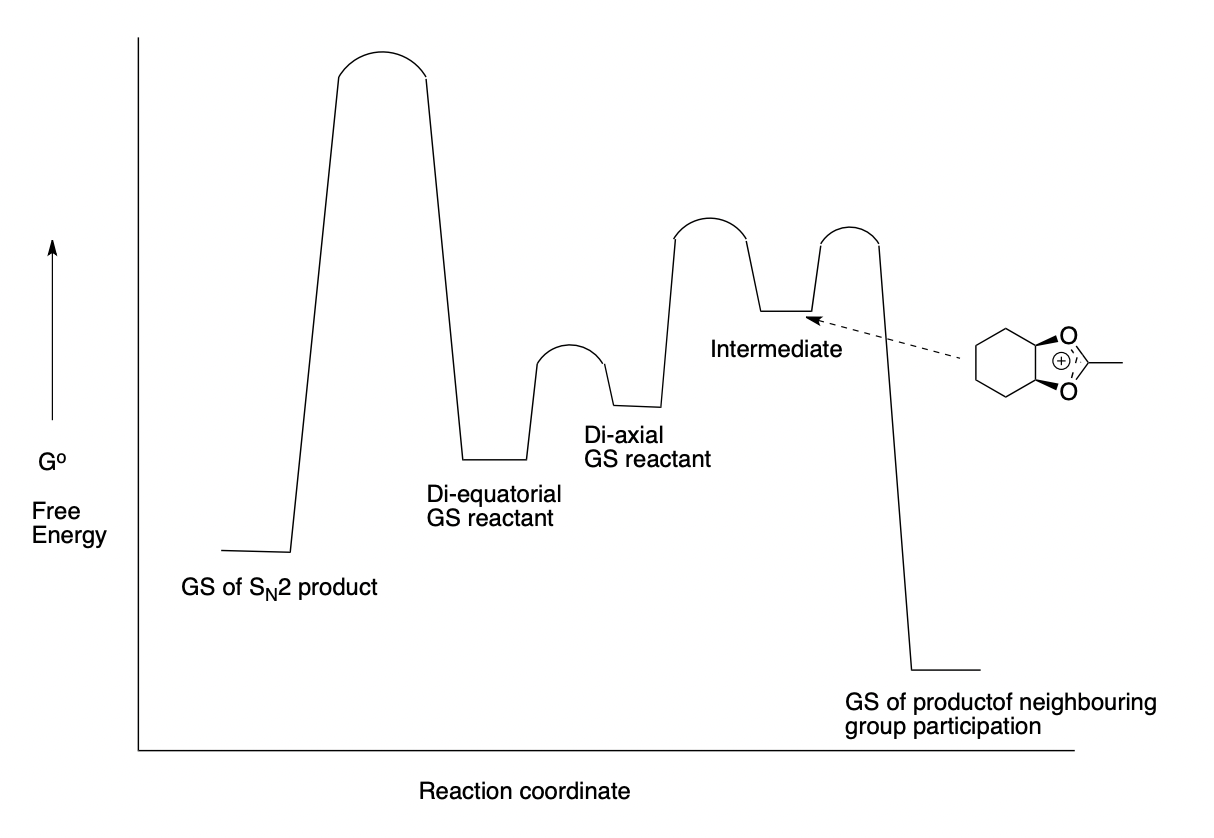
describe neighbouring group participation (= anchimeric assistance)
(strained) cyclic intermediate increases reaction rate
sometimes lead to rearrangement
sometimes affects stereochemistry of products
= intramolecular nucleophilic catalysis (i.e. iodide nucleophilic catalysis)
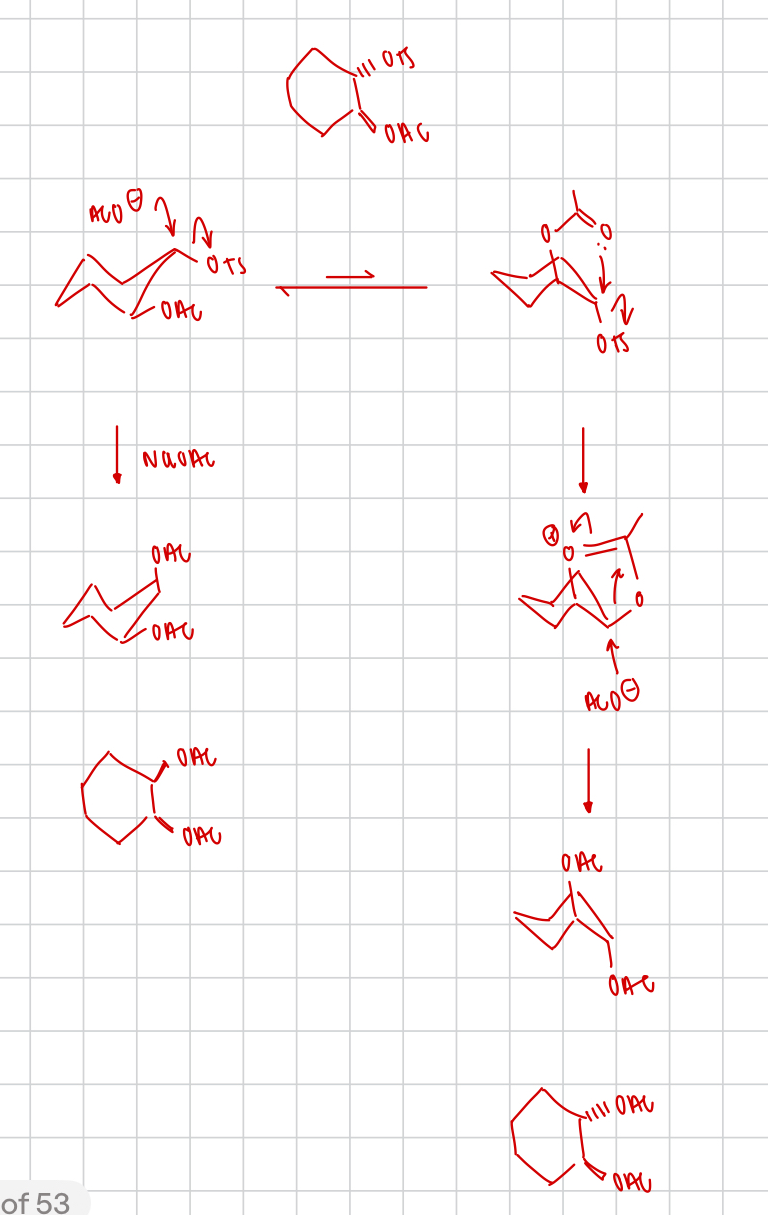
describe the effect of neighbouring group participation here
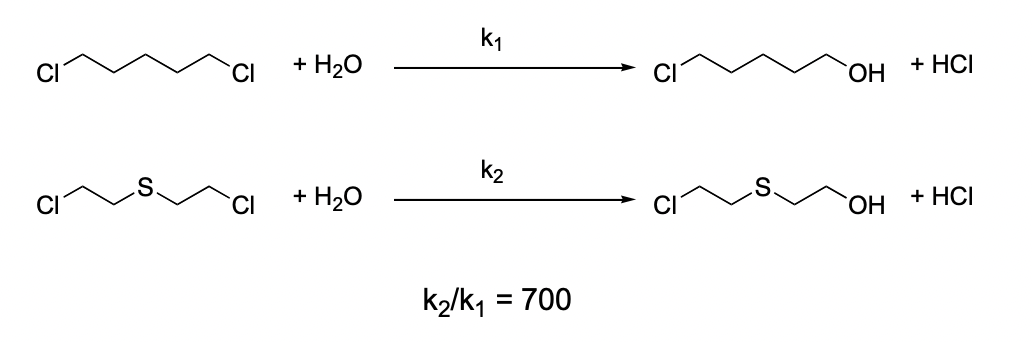
3-exo-tet cyclisation
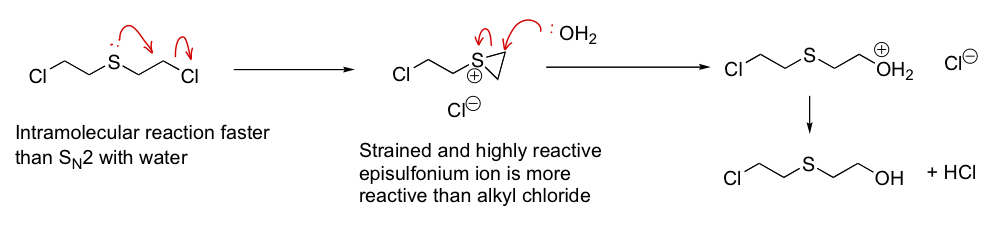
draw the mechanism of this reaction
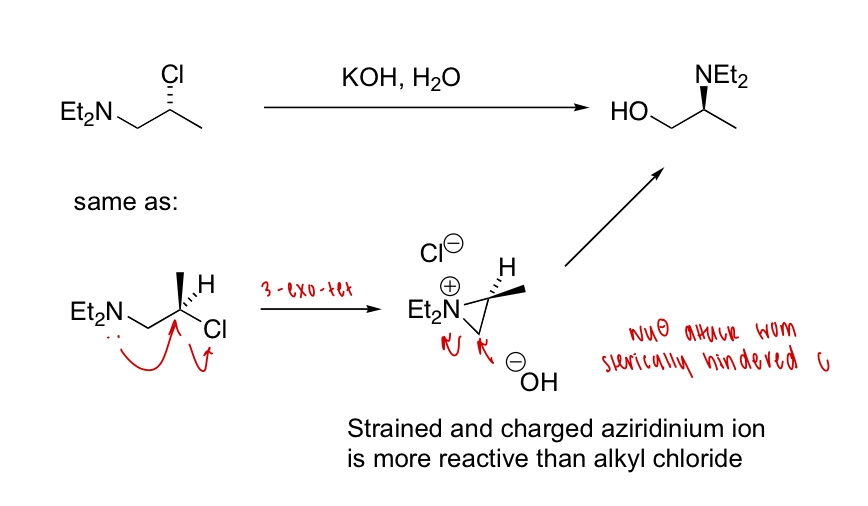
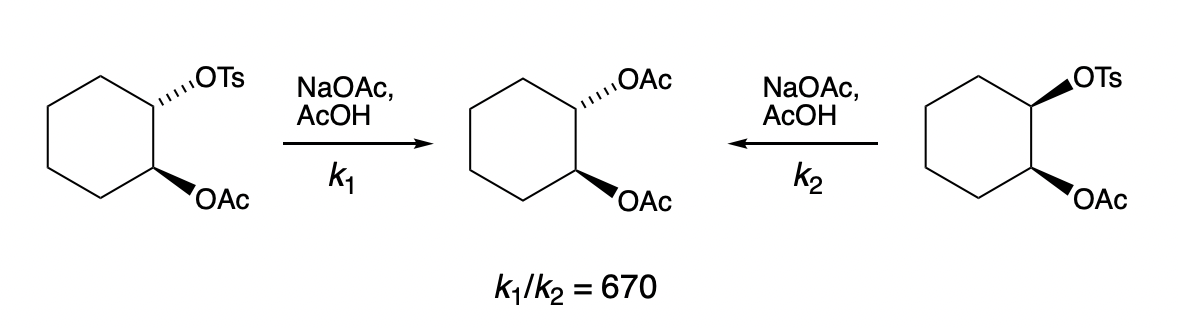
explain why the anti diastereomer is faster than the syn diastereomer
= neighbouring group participation
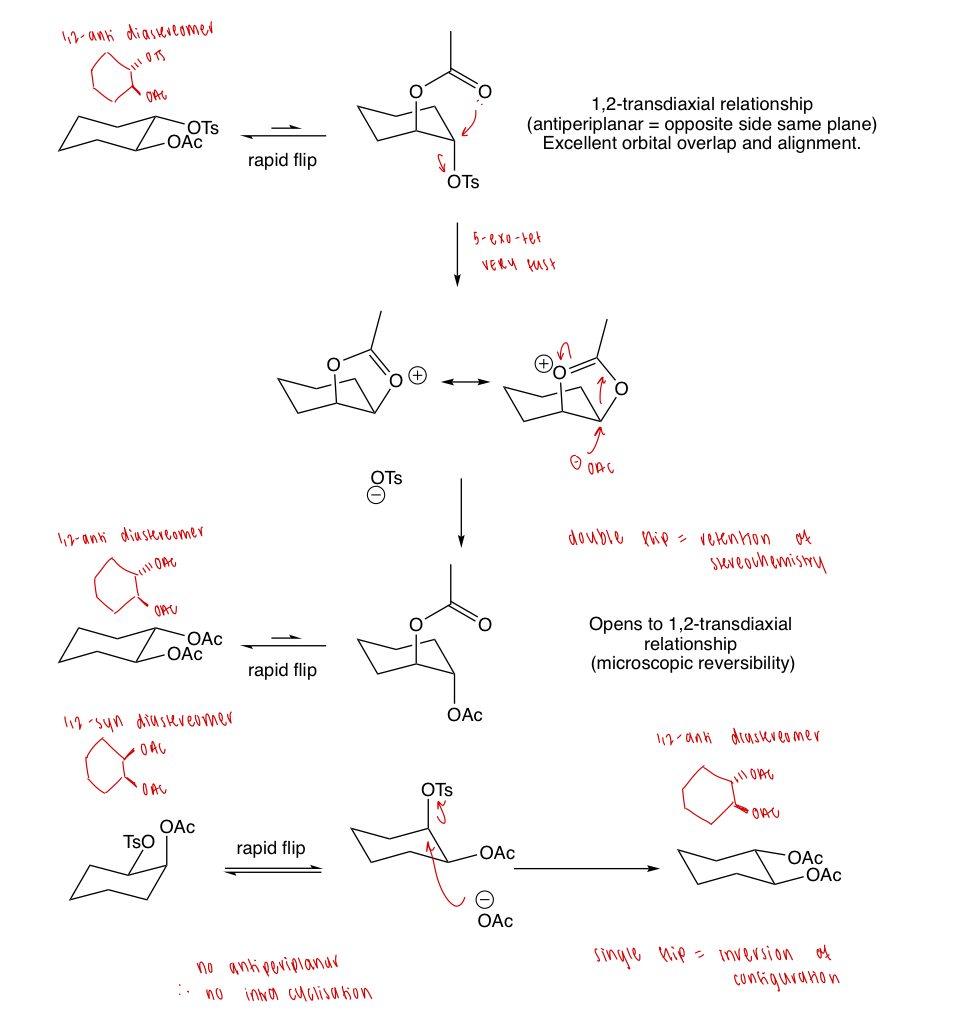
explain the Curtin Hammett principle
the ratio of products formed from one starting material present in two rapidly equilibrating conformations depends only on the energy difference between the two transition states
draw the two pathways of this reaction
LEFT IS WRONG = NEEDS TRANSDIAXIAL
describe SN1 reactions
unimolecular; first order
stepwise; 2 step
loss of stereochemistry