Biochem 285 Week 6
1/27
Earn XP
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
28 Terms
Heterologous gene expression
The process of expressing genes in another organism (host) that doesn’t naturally express those genes so that we can get a particular protein made. We do this to isolate proteins using recombinant DNA.
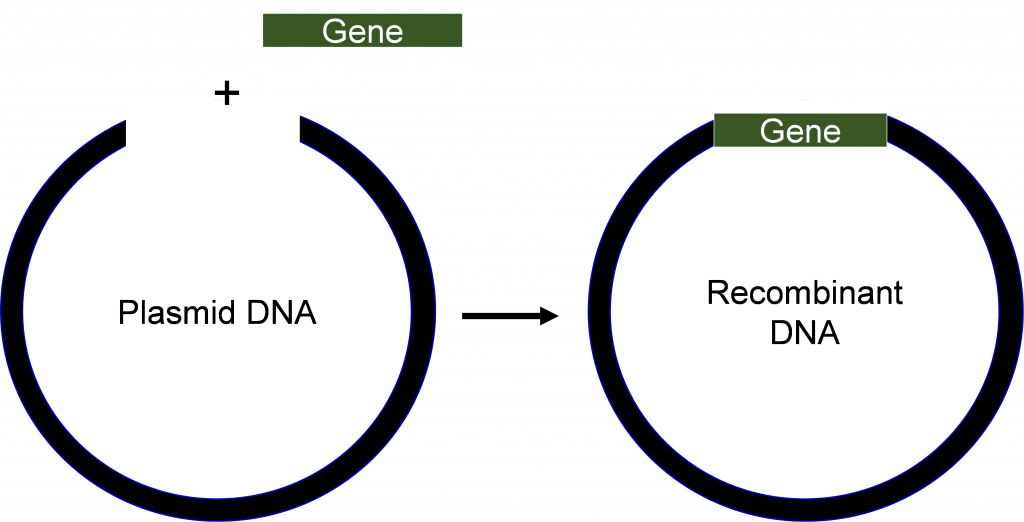
Recombinant DNA
DNA that has been manipulated so it can be expressed

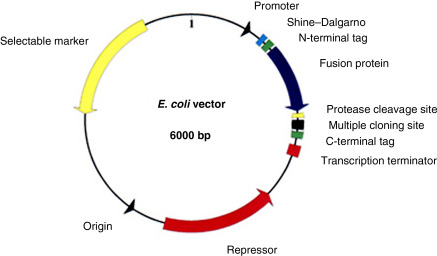
Plasmid (vector)
Bacteria have simplistic circular genomes, with all their genes on one chromosome. They also got their little friend that is an extracellular/extrachromosomal element that holds just a few genes.

When plasmids are use for heterologous gene expression, what is necessary on them? What is not necessary, but is still usual to find?
They contain features that are necessary for gene expression (promoter, ribosome binding site, start/stop codons), for promoting retention of it in the host organism (selectable marker), and allow for replication (origin of replication), and an open reading frame (aka coding sequence/gene). The ORI is not essential for gene expression (along with the selectable marker), but it will be needed for the plasmid to replicate itself so it is still usually found on one. If a plasmid doesn’t have an ORI it’s called a suicide vector. (The selectable marker is a gene sequence that provides antibiotic resistance which stops the host’s immune system from attacking the plasmid.)
Transformation
Process that moves plasmid DNA into host cell. The most common host cell studied is E. coli
Tagging
This is a modification done on the end a gene (C-terminus) which adds a sequence of amino acids (usually a polyhistidine) which can be recognized by an antibody or ion. This is done so it can isolated and purified later in lab using nickel column chromatography! (don’t need to know the italicized part specifically, its just cool).
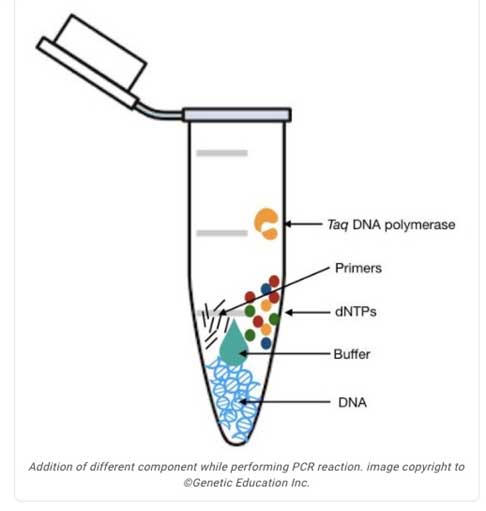
PCR (Polymerase Chain Reaction)
A way we can amplify a specific DNA sequence from an existing template.
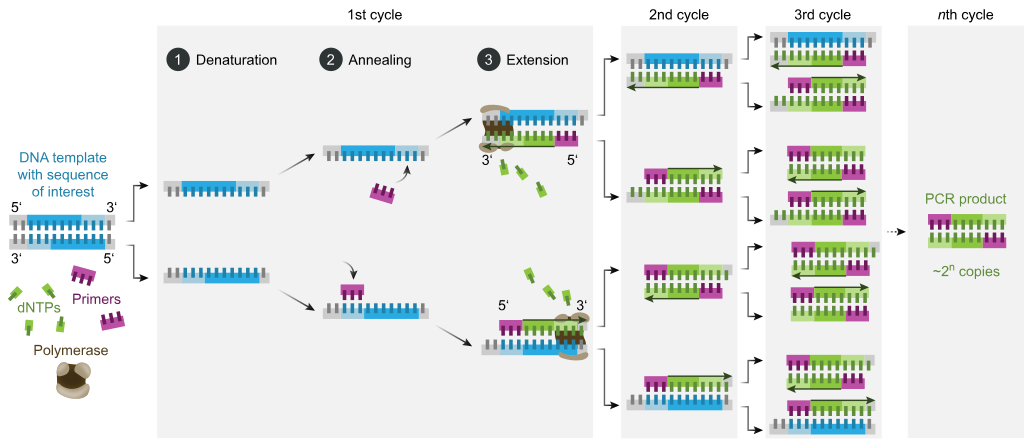
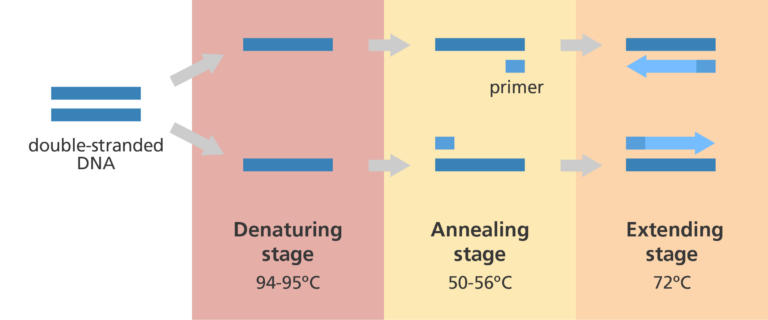
Denaturation (first step of PCR)
We need to somehow separate the strands from the template DNA. So we’ll use heat and crank that thermostat up to 95°C so that the hydrogen bonds that pair the bases are broken. This takes about 30 seconds.

Why don’t we just use helicase to separate the DNA strands in PCR like we do in DNA replication?
To use helicase there now needs to be an origin of replication, helicase also runs on a lot of ATP, and none of this would happen without a signal from cell cycle control to fire the origin in the first place. TL;DR: Too much fucking work. We tryna keep this as simple as possible, so we just use heat.
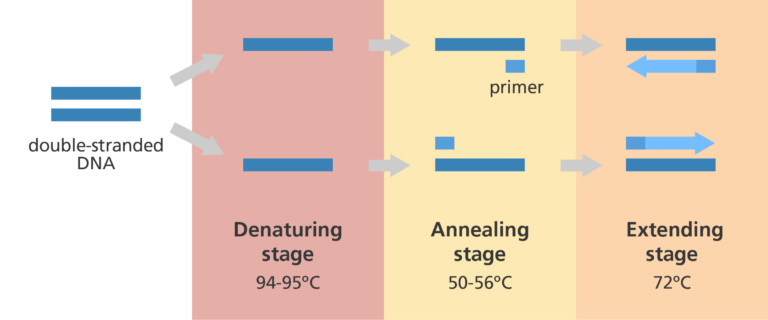
Annealing (second step of PCR)
The strands are separated now. What do we need to do now to start DNA synthesis? Add primers. But first we got to cool things off to 55-60C to allow the short primers to bind to complementary sequences on the template DNA (allowing hydrogen bonding again). We design these primers specifically so that they’ll base pair with the beginnings and ends of the gene. Remember: DNAP can’t start added bases without being primed.
Why don’t we reduce the temperature to even cooler than 55-60°C if we wanna make sure that the primers can hydrogen bond with the bases?
If we reduce the temperature too much in the annealing stage, this will allow for the DNA strands to re-neal, as in join back together into a double strand! All our hard work down the drain! So we reduce the temperature just enough that the primers can bind.

Elongation (third step of PCR)
This is when we increase the temperature (68°C) to one that is optimal for DNAP to do its thing. dNTPs will now be added, elongating the new strands being made. This step can vary in time depending on how long the gene is.
What DNAP is used in the elongation step?
Taq Polymerase because it is a heat tolerant (thermostable) polymerase. We got to keep the heat up to keep the strands from renealing. This polymerase is native to hot springs.
Fidelity
An enzymes ability to replicate DNA with minimal errors. High fidelity enzymes like Taq polymerase have good editing functionality (but there are ones being engineered to be better). Low fidelity polymerases exist and are used on purpose to introduce changes in the DNA sequence.
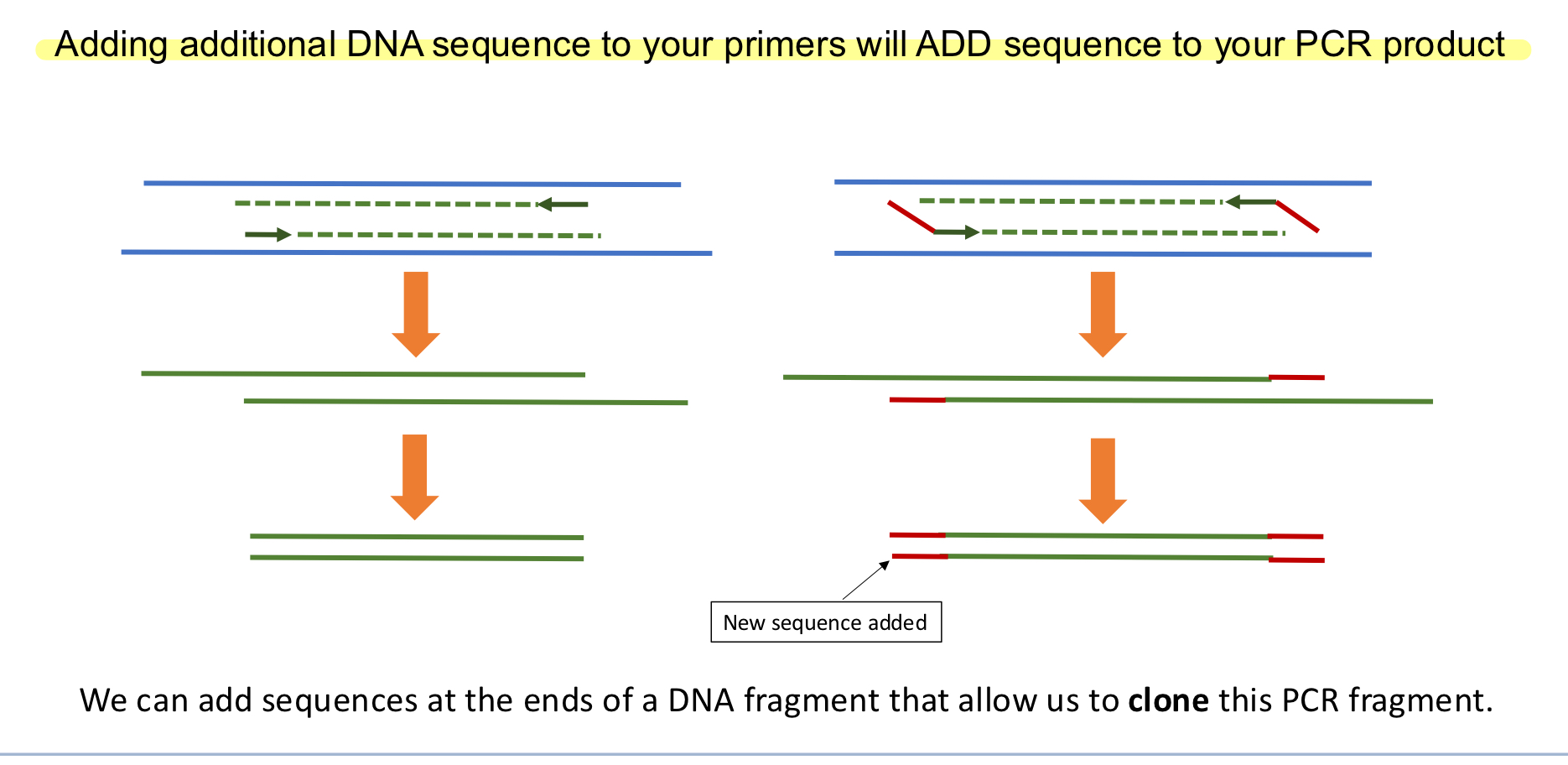
What can be added to PCR primers to modify the final product?
Additional DNA sequences. These would be added to the 5’ end of each primer so that it doesn’t disrupt the gene sequence as it would if you attached it to the 3’ end.
Molecular Cloning of DNA
These are techniques used to insert a DNA fragment of interest into a vector. Vectors are replication machines and can make multiple clones of a DNA fragment, so we insert a fragment of DNA making them recombinant DNA. Note: All vectors that are used for cloning will obviously need an ORI, or else how will they replicate?
Cloning step one
Obtain the DNA (vector + fragment). You can buy ‘em or make your own.
Cloning step two
Cut the DNA. The DNA must be cut so that the vector and fragment can come together. Enzymes do this by cutting them in a way that makes them compatible: leaving ‘em with sticky ends that will anneal
Cloning step three
Paste the DNA. This makes sure that the two separate DNA molecules are now one by connecting phosphodiester bonds through the backbone.
Restriction Enzymes
These endonucleases cleave/cut a phosphodiester bond within a DNA backbone by recognizing palindromic DNA sequences. By cutting in a zigzag along these palindromic sequences, it makes the ends “sticky” (a DNA overhang that can complementarily bind to end of a vector or fragment). There are both sticky end cutters and blunt end cutters (they cut in straight line vs. zigzag). They originate from prokaryotes.
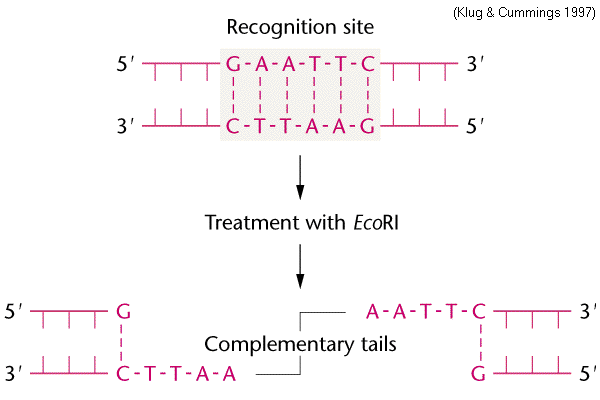
What causes the sticky ends of the DNA vector and the DNA fragment to anneal?
Hydrogen bonds (its just what normal base pairing uses too!)
After the third step in cloning (pasting), what enzyme comes in to seal the nicks in the phosphodiester bond? (hint: it’s not needed in PCR)
Ligase
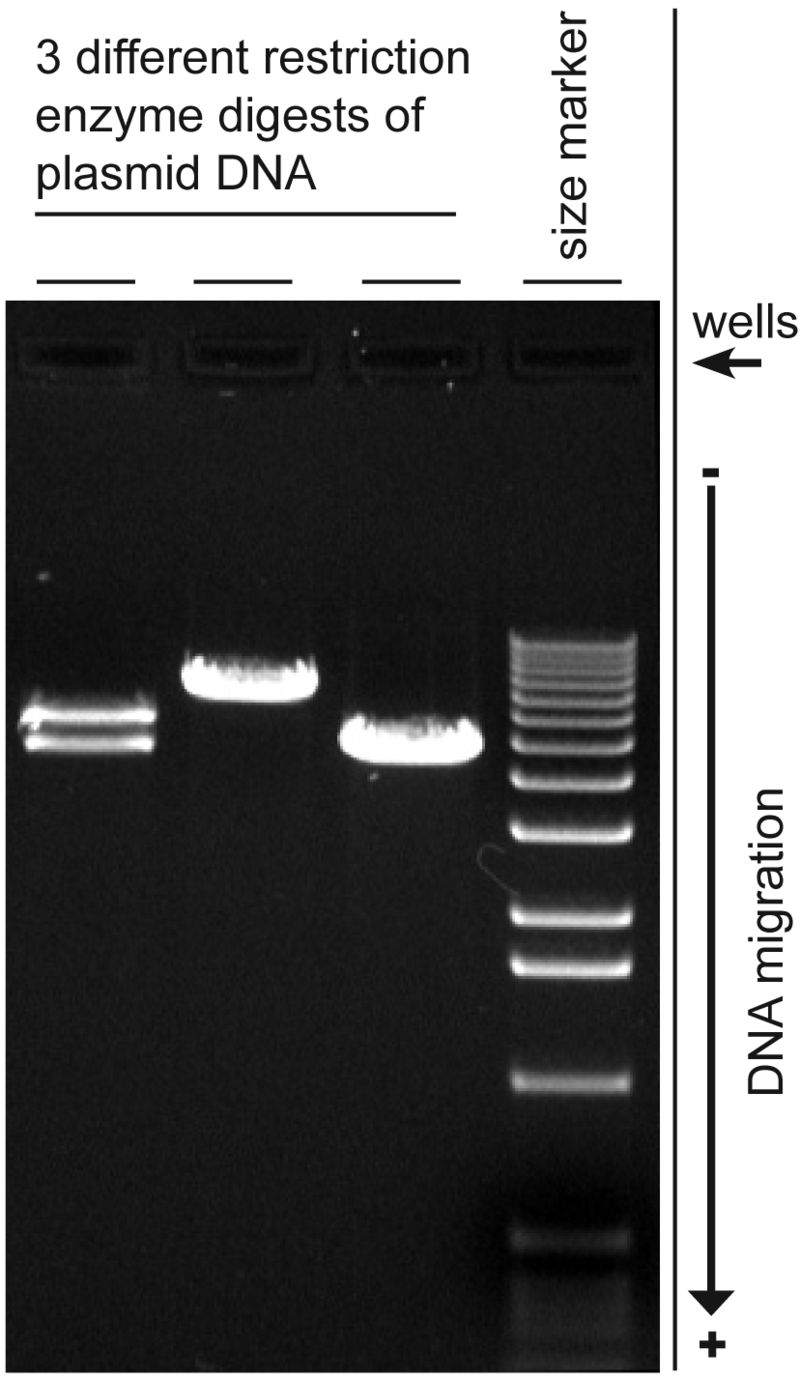
Agarose Gel Electrophoresis
A technique that separates DNA fragments by size. We will use those restriction enzymes (the blunt end cutters) to create linear fragments of the gene of interest off a vector. This is because when we run the DNA through the agarose gel, if it is circular, the gel cannot properly size it. When they’re cut we can run them through the gel, which uses an electrical current, and compare sizes. An uncut sample is also added to see what it would look like if the restriction enzymes failed. The higher up it is on the gel, the larger the fragment is.
Gibson Assembly
An alternate way to clone which allows for multiple DNA fragments to be joined in one step.
What enzymes would give you the digestion pattern in an agarose gel?
Enzymes that are dispersed through different parts of the construct and have unique sizes that will show up well on a gel. Enzymes that are too close together will be too small for the gel (~100bps) as they’ll get washed out.
What can’t restriction digest and agarose gel electrophoresis tell you?
If mutations were introduced during PCR. They can only tell you the size and how close together they were in the vector.
DNA sequencing (Sanger sequencing)
This happens post-PCR, after the DNA has be isolated. It has similar steps to PCR: first step is to heat it up so that strands separate. Then the temperature is lowered just enough so that the primers can anneal. Temperature is back up so that Taq polymerase can work. Now, it keeps adding bases until terminator bases (ddNTPs) are added. These bases are chemically altered so that no more bases can be added. They are also fluorescent and work to tag the end of the fragment. Each terminator base (A,C,G,T) is labeled with a different color so when electrophoresis is done, they can identified by a camera. Convert the colors to letters, and we have a sequence of our piece of DNA!
How can eukaryotic genes be cloned for expression in a prokaryote?
An enzyme (reverse transcriptase PCR) can be used to convert the mRNA of the gene into cDNA (complementary DNA).