Ch 21 - Alpha Carbon Chemistry: Enols and Enolates
1/49
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
50 Terms
alpha position
directly adjacent to a subject position
e.g. in relation to a carbonyl, the positions right next to the carbonyl (not the carbonyl itself)
designated by an α (alpha) symbol
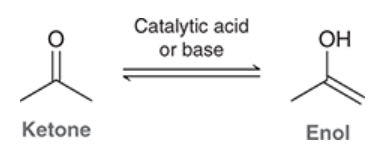
enol
exists in equilibrium with a ketone in the presence of catalytic acid or base
the tautomer of a ketone
generally not favored at equilibrium, unless it is stabilized
very reactive; alpha position is very nucleophilic (lone pair with negative charge)
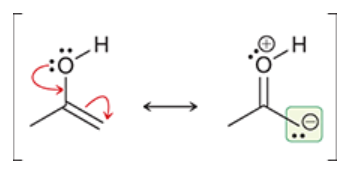
tautomers
rapidly interconverting constitutional isomers that differ from each other in the placement of a proton and the position of a double bond
note: NOT the same as resonance structures
e.g. ketones and their respective enols
tautomerization is catalyzed by trace amounts of acid or base, and is difficult to prevent
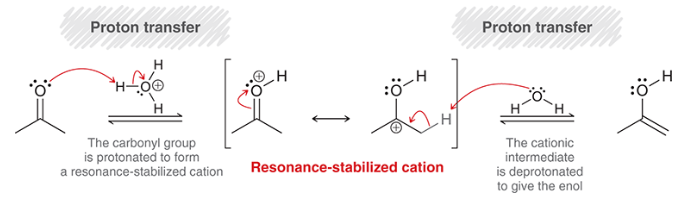
mechanism for acid-catalyzed tautomerization
proton transfer (protonation of carbonyl)
rearrangement of resonance-stabilized cation
proton transfer (deprotonation at alpha position)
mechanism for base-catalyzed tautomerization
proton transfer (deprotonation at alpha position)
rearrangement of resonance-stabilized anion
proton transfer (protonation of anion)
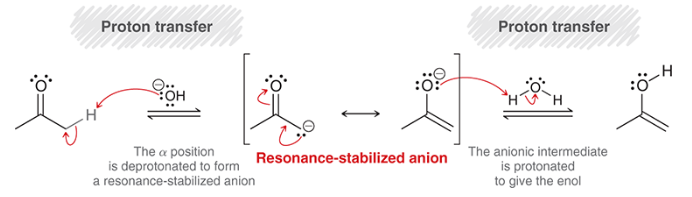
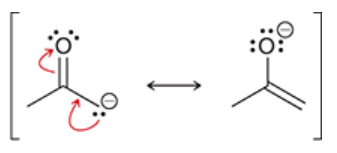
enolate
a resonance-stabilized intermediate that results from deprotonation of a ketone’s alpha position when treated with a strong base
possess two nucleophilic sites which can each attack an electrophile (thus, called ambident nucleophiles) (oxygen or carbon)
O bears the majority of negative charge, but C is more common to attack
more useful than enols because they possess a full negative charge (are more reactive), and can be isolated and stored for short periods of time
most reactions in this chapter will proceed via this intermediate
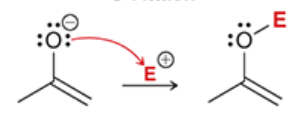
O-attack
when the oxygen atom of an enolate attacks an electrophile
less common than C-attack
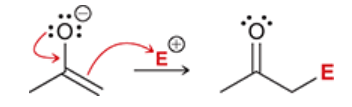
C-attack
when the negative carbon atom of an enolate attacks an electrophile
more common than O-attack
note: when drawing the mechanism, draw the resonance charge with the negative charge on O, even though C is attacking (because O bears more negative charge than C)
typical pKa range of aldehydes and ketones
16-20
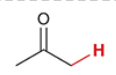
pKa of acetone
19.2
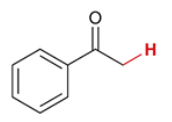
pKa of acetophenone
18.3
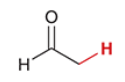
pKa of acetaldehyde
16.7
equilibrium between alkoxide base and ketone/aldehyde
pKa range of aldehydes and ketones is similar to the range of alcohols
when alkoxide ion is used as base, equilibrium is established where both alkoxide and enolate are present (usually there is less of the enolate present at equilibrium)
equilibrium favors the higher pKa
commonly used bases for irreversible enolate formation
sodium hydride (H- is the nucleophile)
LDA (lithium diisopropylamide)
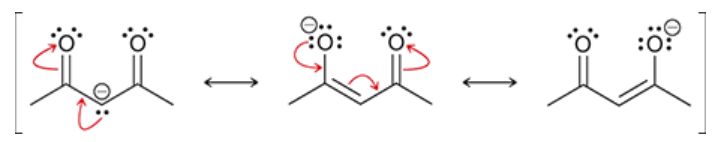
enolate formed by attack of a molecule with two beta carbonyl groups
protons sandwiched between the carbonyls are highly acidic
pKa is generally around 9
acidity is because of the highly stabilized anion formed upon deprotonation
negative charge in anion is spread over 2 oxygens and 1 C
nearly complete enolate formation can be completed with just hydroxide or alkoxide
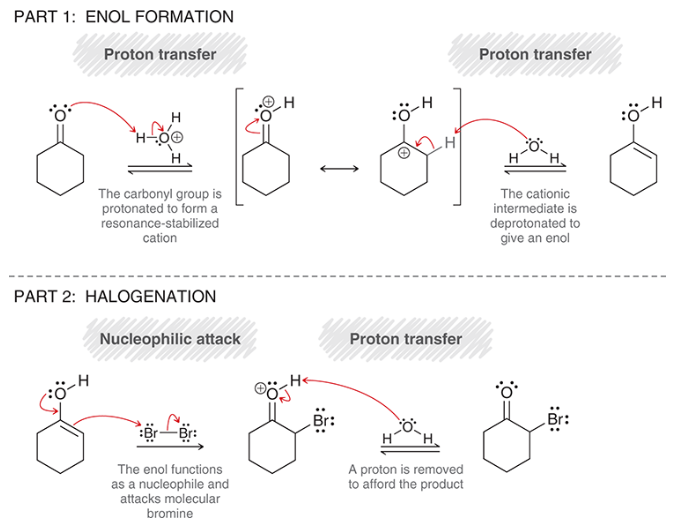
mechanism for acid-catalyzed halogenation of ketones
part 1: enol formation
proton transfer (protonation of carbonyl)
rearrangement of resonance-stabilized cation
proton transfer (deprotonation of alpha position)
part 2: halogenation
nucleophilic attack by enol
proton transfer (deprotonation)
alpha halogenation
undergone by ketones and aldehydes under acid-catalyzed conditions
(does not occur readily with carboxylic acids, esters, or amides)
observed for Cl, Br, and I (not F)
variety of solvents can be used, e.g. acetic acid, water, chloroform, diethyl ether
rate is independent of concentration of identity of the halogen (halogen does not participate in the rate-determining step)
autocatalytic
occurs primarily at the more substituted side of the ketone/aldehyde if the starting material is unsymmetrical
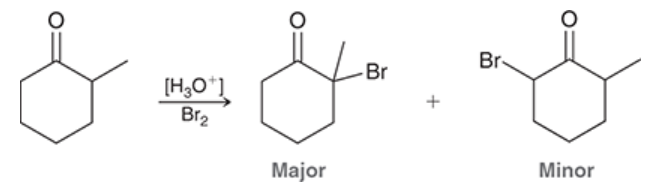
autocatalytic
when the reagent necessary to catalyze a reaction is produced by the reaction itself
e.g. alpha halogenation; HBr is a byproduct, which is an acid and is capable of catalyzing the enol formation
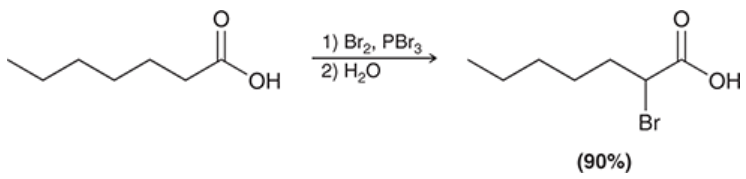
Hell-Volhard-Zelinsky reaction
alpha halogenation of a carboxylic acid when treated with bromine in the presence of PBr3
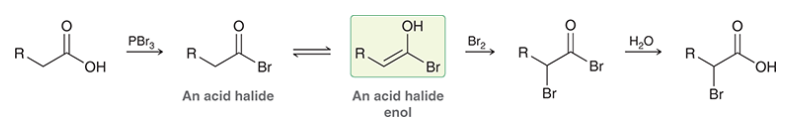
sequence of Hell-Volhard-Zelinsky reaction
carboxylic acid reacts with PBr3 → forms acid halide
acid halide exists in equilibrium with enol
enol functions as nucleophile, undergoes halogenation at alpha position
hydrolysis regenerates carboxylic acid
haloform reaction
reaction in which a methyl ketone is converted into a carboxylic acid upon treatment with excess base and excess halogen, followed by aqueous acid
most efficient when the other side of the ketone has no alpha protons
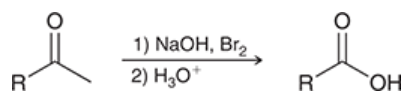
(believed) mechanism for haloform reaction
alpha protons are removed and replaced with Br atoms, one at a time
tribromomethyl group functions as a leaving group → results in nucleophilic acyl substitution
resulting carboxylic acid is deprotonated → produces carboxylate and CHBr3 (bromoform)
formation of carboxylic drives the reaction to completion
(note: if performed with Cl or I instead of Br, chloroform or iodoform is the by-product)
reaction is followed by treatment with a proton source (acid) to protonate the carboxylate and form carboxylic acid
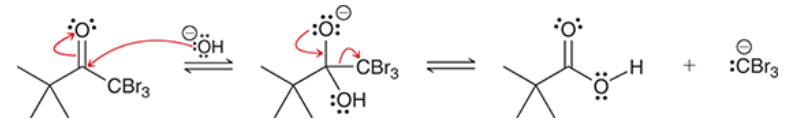
aldol addition
an aldehyde or ketone is attacked by enolate
product is (always) a beta-hydroxy aldehyde or ketone
product exhibits both aldehydic and hydroxyl groups
for most simple aldehydes, equilibrium favors aldol product
for most ketones, aldol product is not favored; poor yields are common
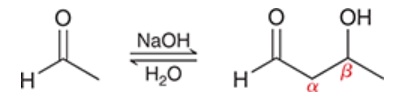
mechanism for aldol addition
proton transfer (deprotonation of alpha position to form enolate)
nucleophilic attack by enolate (attacks aldehyde)
proton transfer (protonation of alkoxide)
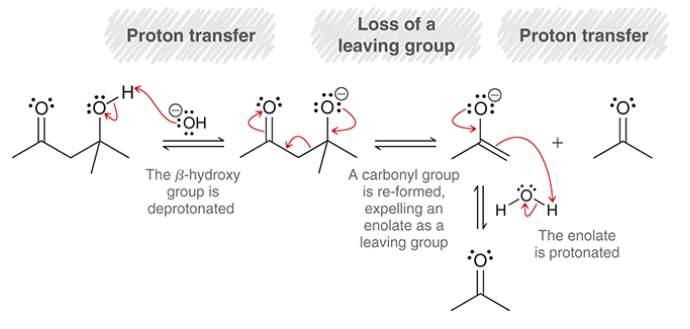
retro-aldol reaction
reverse process of aldol addition
beta-hydroxy ketone/aldehyde is converted back into a ketone/aldehyde
mechanism for retro-aldol reaction
proton transfer (deprotonation of beta-hydroxy group)
loss of leaving group (enolate is expelled)
proton transfer (protonation of enolate)
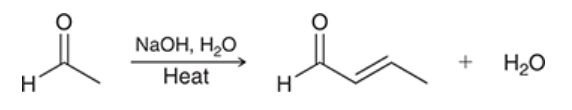
aldol condensation
reaction in which the product of aldol addition is heated in acidic or basic conditions and undergoes elimination to produce unsaturation between alpha and beta positions
two steps: aldol addition plus dehydration
condensation refers to the addition of two molecules accompanied by the loss of a small molecule, e.g. water, CO2, or N2
product is an alpha,beta-unsaturated ketone/aldehyde
if two stereoisomeric pi bonds can be formed, the product with fewer steric interactions is generally the major product
yields are generally much greater than yields for addition reactions
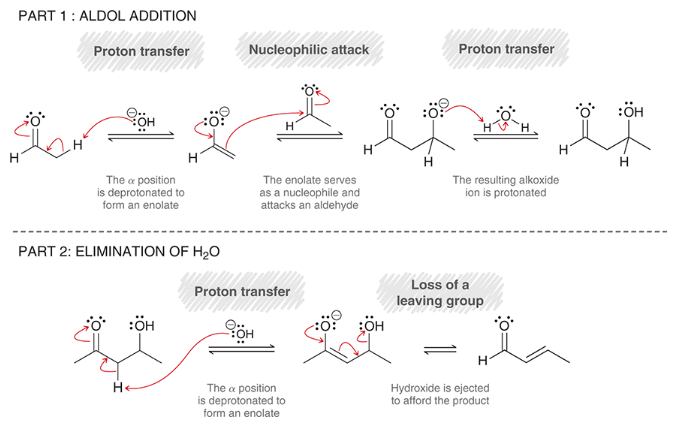
mechanism for aldol condensation
part 1: aldol addition
proton transfer (deprotonation to form enolate)
nucleophilic attack (enolate attacks aldehyde)
proton transfer (protonation of alkoxide)
part 2: elimination of H2O
proton transfer (deprotonation of alpha position)
loss of leaving group (hydroxide is ejected)
E1cb mechanism
an elimination reaction in which the leaving group only leaves after deprotonation occurs
occurs at the end of aldol condensation
cb stands for conjugate base
1 indicates that the reaction is first order
crossed aldol (aka mixed aldol) reaction
an aldol reaction that can occur between different partners
e.g. two starting materials may be treated with a base and produce four different aldol products
of little practical use
only efficient if they can be performed in a way that minimizes the number of possible products
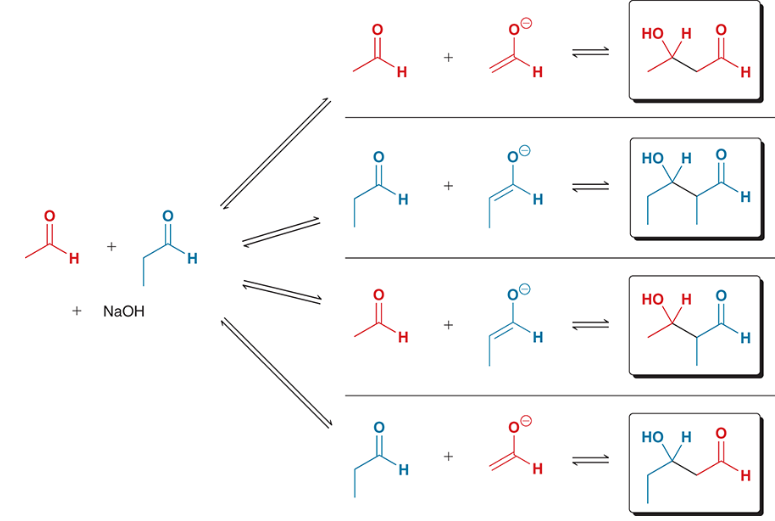
ways to minimize the number of possible products in a crossed aldol reaction
1) one of the aldehydes lacks alpha protons and possesses an unhindered carbonyl group (e.g. formaldehyde)
2) LDA is used as a base (causes irreversible enolate formation)
directed aldol addition
a technique for performing a crossed aldol addition that produces one major product
success is limited by the rate at which enolate ions can equilibriate (function as a base and deprotonate a molecule of aldehyde)
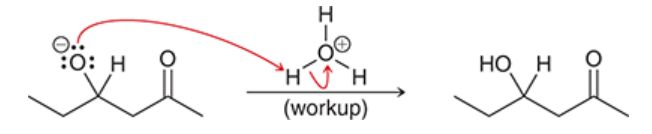
intramolecular aldol reactions
may occur in compounds that possess two carbonyl groups
one end forms an enolate group, which attacks the carbonyl group
preference is for formation of five- and six-membered rings
(smaller rings are possible but not generally observed)
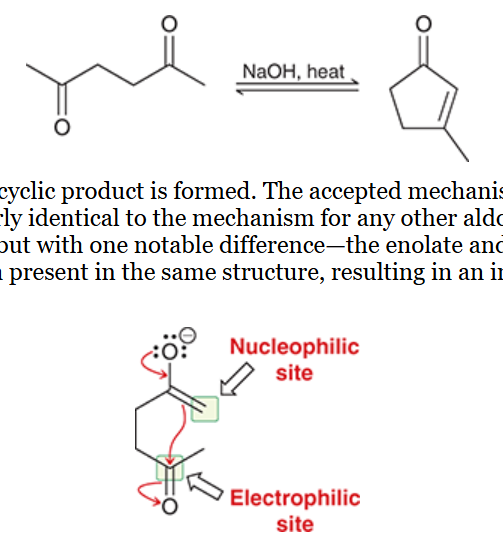
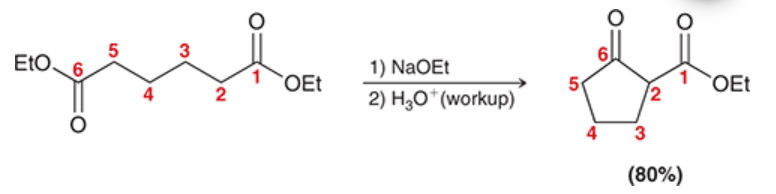
Claisen condensation
the reversible condensation of an ester
nucleophilic acyl substitution reaction in which the nucleophile is an ester enolate, electrophile is an ester
product is a beta-keto ester
ester is treated with 1) NAOEt and 2) H3O+ (workup)
differs from aldol reaction in the fate of the tetrahedral intermediate; tetrahedral can expel leaving group to reform carbonyl
hydroxide can NOT be used as base because it can cause hydrolysis of the starting ester
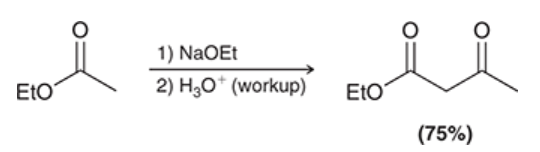
mechanism for Claisen condensation
proton transfer (deprotonation)
nucleophilic attack (enolate is nucleophile, attacks ester)
loss of leaving group (alkoxide ion gets ejected)
proton transfer (protonation of alpha position)
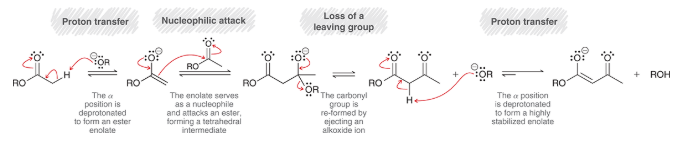
crossed Claisen condensation
a Claisen condensation that occurs between two different partners
produce a mixture of products, just like crossed aldol reaction
only efficient if one of the two criteria are met:
1) one ester has no alpha protons and cannot form an enolate
2) LDA is used as base to irreversibly form ester enolate (which then gets treated with a different ester)
Dieckmann cyclization
intramolecular Claisen condensation
product is a cyclic beta-keto ester
one carbonyl is the ester enolate (nucleophile), the other carbonyl is the electrophile
preference is for formation of five- and six-membered rings, just like intramolecular aldol reactions
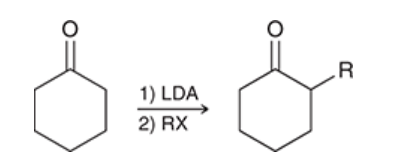
alpha alkylation
alpha position of a ketone or aldehyde via formation of an enolate, followed by treatment of enolate with an alkyl halide
enolate functions as nucleophile, attacks alkyl halide in Sn2 fashion (usually restrictions for Sn2 apply)
choice of base is important: hydroxide or alkoxide ions cannot be used. stronger base, e.g. LDA, can be used
if ketone is unsymmetrical, two possible enolates can be formed (thermodynamic and kinetic)
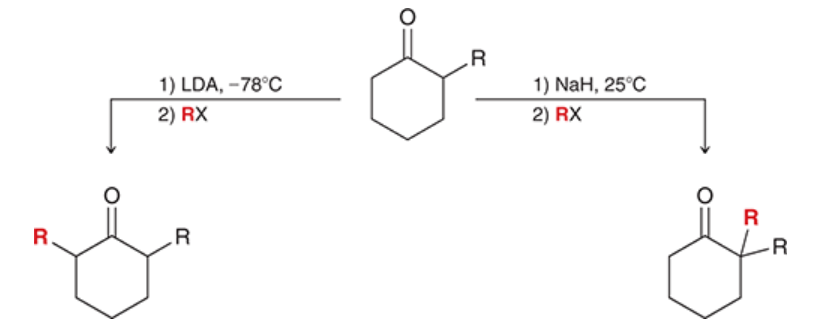
how to choose the formation of either a thermodynamic or kinetic enolate
if kinetic enolate is desired, use LDA at low temperature (-78*C)
if thermodynamic enolate is desired, nonsterically hindered base (e.g. NaH) can be used at room temperature
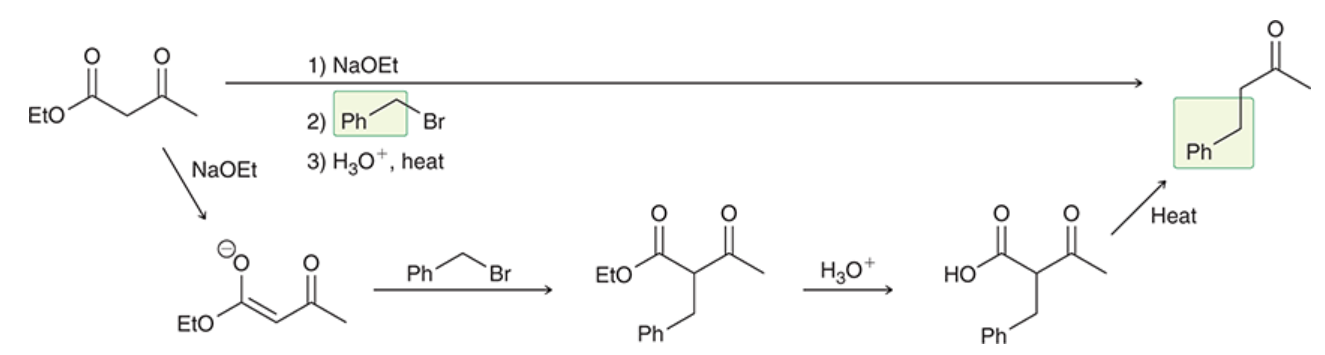
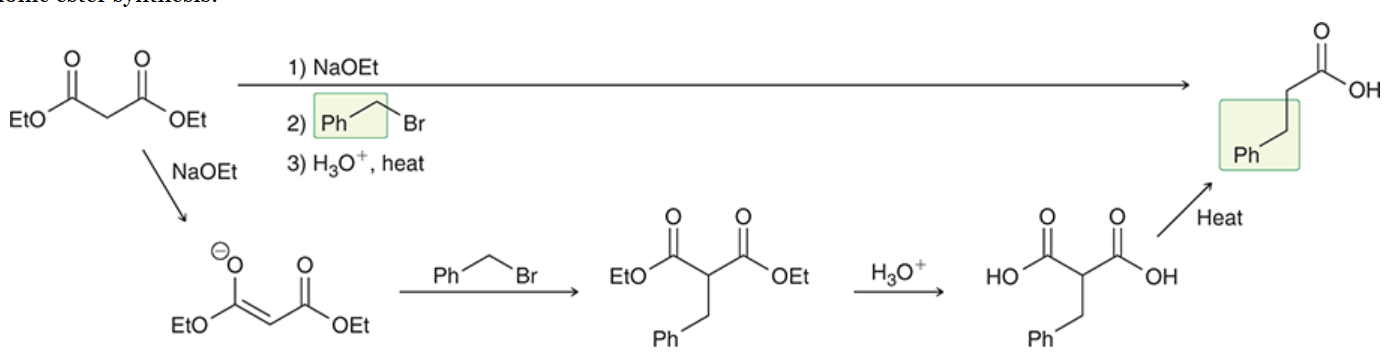
acetoacetic ester synthesis
a three-step synthesis that converts an alkyl halide into a methyl ketone with the introduction of three new carbon atoms
(prepares substituted derivatives of acetone)
a way to avoid the issue of polyalkylation
ethyl acetoacetate gets deprotonated by strong base → forms highly stabilized enolate
enolate is alkylated by alkyl halide (via Sn2)
alkylated product is treated with aqueous acid, results in hydrolysis of ester group
if hydrolysis is performed at elevated temp, resulting carboxylic acid undergoes decarboxylation to produce a monosubstituted acetone derivative and CO2
malonic ester synthesis
an efficient method for creating substituted derivatives of acetic acid
enables the transformation of a halide into a carboxylic acid with the introduction of two new carbon atoms
diethyl malonate is deprotonated, then the resulting enolate is treated with an alkyl halide (best to use primary alkyl halide)
alkylated product is treated with aqueous acid, both ester groups are hydrolyzed to give a diacid
if hydrolysis is performed at elevated temp, diacid will undergo decarboxylation to produce an acetic acid derivative and CO2
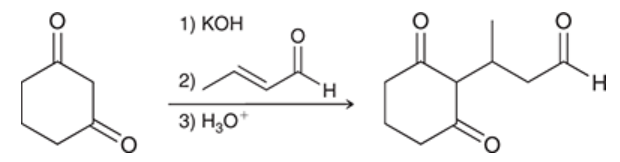
Michael reaction
reaction in which a nucleophile attacks a conjugated pi system
results in a 1,4-addition (aka conjugate addition)
a diketone is deprotonated to form a highly stabilized enolate ion
the enolate serves as a nucleophile in 1,4-addition
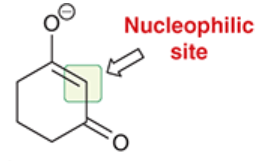
Michael donor
the highly stabilized enolate in a Michael reaction
the nucleophile
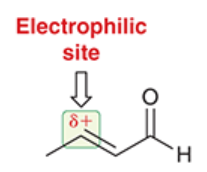
Michael acceptor
the alpha,beta-unsaturated aldehyde in a Michael reaction
the electrophile
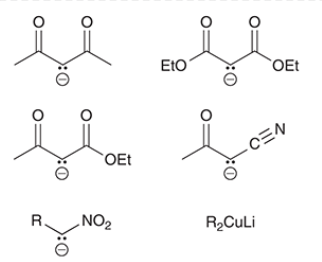
common Michael donors
only highly stabilized enolates
not regular enolates
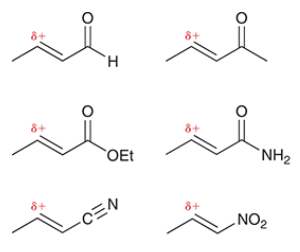
common Michael acceptors
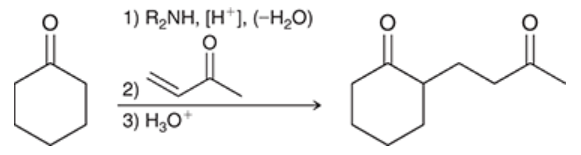
Stork enamine synthesis
a type of Michael reaction in which an enamine is the nucleophile
ketone is converted into enamine by treatment with secondary amine
enamine reacts with a suitable Michael acceptor and generates an intermediate that is both iminium ion and enolate ion
treated with aqueous acid, both groups get converted into carbonyls
three steps:
1) enamine formation
2) Michael addition
3) hydrolysis
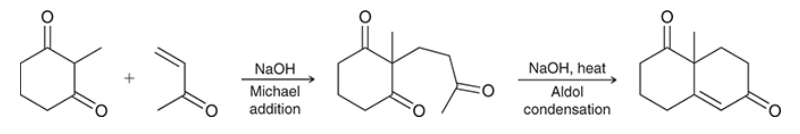
Robinson annulation
two-step method to form a ring in which Michael addition is followed by intramolecular aldol condensation
often used for synthesis of polycyclic compounds
similarities and differences between aldol addition, Claisen condensation, and Stork enamine synthesis
all produce difunctionalized compounds, but the positioning is different
aldol addition and Claisen condensation produce 1,3-difunctionalized compounds
Stork enamine synthesis produces 1,5-difunctionalized compounds
aldol addition produces a carbonyl and a hydroxyl group, Claisen condensation produces an ester and a carbonyl group (oxidation states are different)
alkylation of alpha and beta positions
an enolate formed by Michael addition can be quenched with water to give the product
or the enolate can be treated with alkyl halide → get alkylated at alpha position
both alpha and beta positions can be alkylated in one reaction flask (the two alkyl groups do not need to be the same)