ABDI STUDY GUIDE
1/42
Earn XP
Description and Tags
things from king abdi stidy guide
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
43 Terms
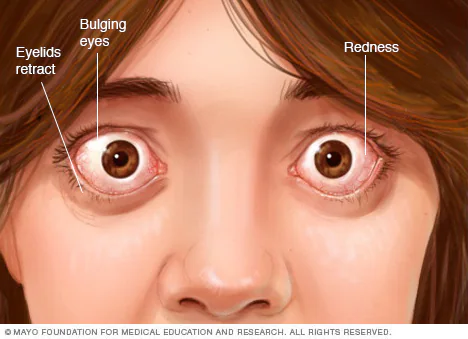
Primary Hyperthyroidism
High fT4 & Low TSH
Most common cause: Grave’s Disease! (anti-TSHR positive [thyroid stimulating hormone receptor])
excess fT4 NOT dependent on hypothalamus-pituitary-thyroid axis
Pituitary Hyperthyroidism (RARE)
TSH normal/high
due to pituitary TSH-secreting tumor (benign)
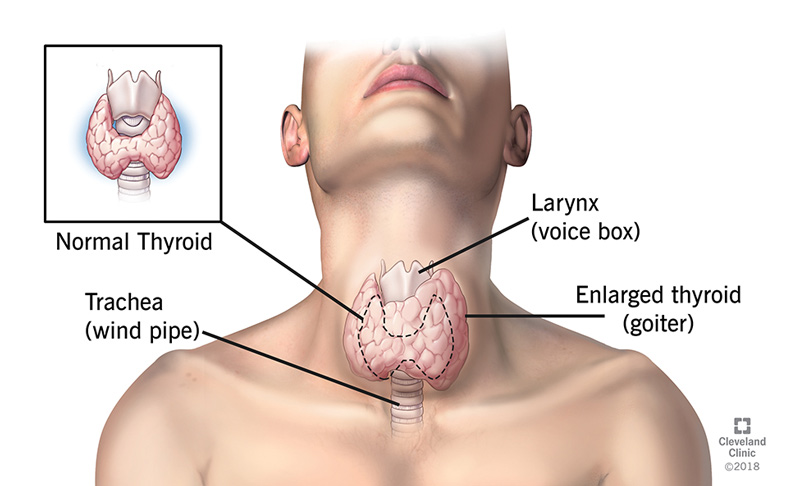
Primary Hypothyroidism
High TSH, Low fT4
Low ft4 NOT dependent on hypothalamus-pituitary-thyroid axis
Most common cause: Hashimoto Dz!
Secondary Hypothyroidism
Low/Normal TSH, low fT4
Hypopituitarism (pituitary gland not making enough hormones b/c of adenoma/radiation/destruction of pituitary
Cushing Syndrome
persistent hypercortisolism (prolonged exposure to cortisol)
Dx:
(+) 24hr UFC (urine free cortisol): 3x abv normal
overnight low dose DST: ≥10μg/dL highly suggestive (<1.8 strongly rules out Cushing syndrome)
Primary caused by adrenal adenoma/carcinoma or bilateral adrenal hyperplasia
“moon face”
iatrogenic: most common cause of glucocorticoild admin
Cushing Dz: pituitary adenomas
Ectopic ACTH: lung cancer, pancreatic cancer
Investigation of high cortisol secretion mechanisms
ACTH-dependent: 8am cortisol >15ug/dL = almost always pituitary gland
Overnight High dose DST to differentiate from ectopic: 8am cortisol <5ug/dL, or suppression is 50% or more vs baseline, source is pituitary)
ACTH-independent: high dose glucocorticoids administration should be excluded first. issue is with adrenal gland itself
Addison Disease
Low 8am aldosterone/very small increase in cortisol lvl after cosyntropin (synthetic form of ACTH) stimulation
>18 ug/dL is indicative of normal adrenal function
Primary Addison disease caused mainly by an autoimmune mechanism
Conn Syndrome
Screening: Ratio of plasma aldosterone conc. to plasma renin (enzy from kidneys) activity, must be confirmed by 24h urinary aldosterone level
Primary hyperaldosteronism, caused by adrenal adenoma or bilateral adrenal hyperplasia; adrenal glands produce too much aldosterone.
Diabetes insipidus neurogenic (BRAIN)
Central DI: insufficient secretion of ADH by posterior pituitary gland (base of brain)
Polyuria, low urine osmolality
Overnight water deprivation test: patient not able to concentrate urine during deprivation but can after ADH injection!
Diabetes insipidus nephrogenic (KIDNEY)
ADH receptor on collecting ducts or distal convoluted tubules not functional
Polyuria, low urine osmolality
Overnight water deprivation test: patient not able to concentrate urine in either case
Syndrome of inappropriate ADH (SIADH)
Low plasma Na, High urine Na (>20mEq/L), urine osmolality >100mOsm
Normal blood volume
Caused by tumors such as small cell carcinoma of the lung, or drug chlorpropamide
Body produces too much ADH (antidiuretic hormone) → body retains too much water → diluting blood & having critically low sodium lvls
GH (Growth Hormone) excess
Is always seen with high IGF-1 (insulin-like growth factor) (normal IGF-1 excludes GH excess)
Indicated by:
Very high GH in a random blood specimen
No response to glucose suppression test of a relatively normal GH patient
Immature menopause
Persistently high FSH (best test) - follicle-stimulating hormone
Ovarian Failure
Persistently high FSH
due to elimination of follicles by pathologic process
Hyperprolactinemia
high plasma prolactin (prolactin is a hormone related to lactation & breast tissue development
Most common cause: prolactinoma (prolactin-secreting tumor)
Causes amenorrhea in women & gynecomastia in men
Gonadal Fxn tests
Females: No progesterone production following hCG stimulation indicates Primary Gonadal Hypofunction
Males: testosterone lvl <150n/dL following hCG stimulation indicates Primary Hypogonadism
FSH ≤10 mU/mL: Hypogonadotropic
FSH: ≥20 mU/mL: Hypergonadotropic
PKU
↑↑in PA (phenylalanine) & its metabolites in serum and urine (mousy odor)
Screened by Guthrie test on blood spots from newborns (bacterial growth is positive result), confirmed by HPLC or MS/MS.
In urine: Phenistix reaction (blue-gray to green); rapid paper-strip urine test to detect elevated lvls of phenylpyruvic acid (by-pdt of phenylalanine)
Mutation in PAH gene encodes phenylalanine hydroxylase
Hyperphenylalaninemia may also happen in BH4 deficiency
Tyrosinosis
High tyrosine and succinylacetone in blood and urine (cabbage-like odor), also tyrosine crystals in urine
In urine: nitrosonaphtol test is positive
MS/MS for confirmation
There are 3 types:
Type I more common and the most severe (cancer risk later in life)
Mutation in FAH gene
Alkaptonuria
Increase in homogentisic acid level in blood & urine
Mutation in HGD gene which encodes homogentisic acid oxidase
Darkening of urine upon standing
Screening test: ferric chloride on urine (turns blue)
MS/MS
MSUD
Accumulation and excretion of α-ketoacids in urine (maple syrup odor)
Modified Guthrie test (inhibitor 4-azaleucine)
Microfluorometric assay
MS/MS
Complete absence or severe deficiency of BCKD enzyme complex
Branched amino acids: valine, leucine, isoleucine
Homocystinuria
Increase in homocysteine and methionine levels
Mutation in CBS gene which encodes cystathionine β-synthase required for methionine metabolism
Modified Guthrie test (inhibitor L-methionine sulfoximine)
Acute inflammation
Negative Acute Phase Reactants:
Albumin
Transferrin
Positive Acute Phase Reactants:
CRP
Haptoglobin
Complement
β2M
Ceruloplasmin
**Increased α1 and α2 on SPE
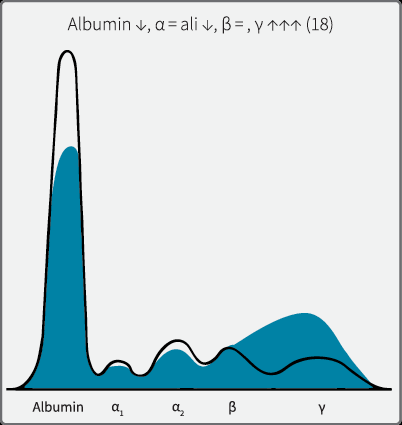
Cirrhosis
SPE pattern: Polyclonal increase in gamma with beta-gamma bridging
Blood-brain barrier damage (meningitis)
CSF electrophoresis: increase on protein
Multiple Sclerosis (MS)
CSF electrophoresis: oligoclonal bands, Myelin basic protein (MBP), sign of myelin damage in general (not specific)
Jaundice: pre-hepatic
increase in unconjugated bilirubin
Amount of bilirubin delivered to liver increased
Most common cause hemolytic anemia
Jaundice: hepatic (d/os of bilirubin transport or metabolism)
Crigler-Najjar syndrome, Gilbert's disease, and neonatal physiologic jaundice of the newborn: Increase in unconjugated bilirubin
Dubin-Johnson syndrome, Rotor syndrome: Increase in conjugated bili
Crigler-Najjar syndrome
rare but serious, risk of kernicterus.
D-J syndrome
excretion into bile defective, dark granules on liver biopsy
Rotor syndrome
clinically similar to D-J syndrome, excellent prognosis, no dark granules on liver biopsy
Alcoholic fatty liver
slight increase in ALT, AST, GGT (Gamma-Glutamyl Transferase)
fatty infiltrates in vacuoles of liver cells on biopsies
mildest form of liver Dz
Alcoholic Hepatitis
mod. increase in ALT, AST, GGT, ALP, total bili >5mg/dL
AST/ALT ratio (De Ritis ratio) >2.0
Albumin reduced; INR increased
Threatening sign: increased creatinine (may precede hepatorenal syndrome and death)
Moderate severity
Alcoholic Cirrhosis
increase in LFTs (ALT, AST, GGT, ALP, total bili), decrease in albumin,
definitive Dz: liver biopsy
last & most severe form :O
Hepatitis B
HBSAg: first to be detected during acute phase, not infectious by itself
HBeAg: second to be detected, with HBSAg only during acute phase, highly infectious.
HBcAb: first Ab to be detected, core window: IgM HBcAb the only marker to be detected, blood could be infective.
HBeAb: second Ab to be detected (may indicate HBeAg loss)
HBSAb: detected late during infection (around 3-6 months after infection), can also be detected years after, indicating clearance of the virus and natural active immunity.
IgG HBcAb: it may be detected with HBSAb which indicates recent infection
Hepatitis C
Positive anti-HCV Ab using ELISA
Real time PCR for virus load
Some patients may clear the virus (Ab+, HCV RNA -)
Myocardial Infarction (MI)
CK-MB: rapidly post-MI (4 to 6 hours) returns to baseline after 2 to 4 days (short window of time after a suspected MI)
LD: levels remain elevated for up to 1 week but are not detectable until 24 to 48 hours post-MI.
Cardiac troponins: detectable in the plasma at 3 to 12 hours after myocardial injury, peaking at 12 to 24 hours and remaining elevated for more than 1 week: 8 to 21 days for TnT and 7 to 14 days for TnI.
Predominant hyperlipidemia
Plasma cholesterol level >200 mg/dL
Related to high LDL (bad cholesterol)
Most common primary cause is Familial hypercholesterolemia (AD deficiency of LDL-R)
Secondary causes: DM, hypothyroidism
Predominant hypertriglyceridemia
Usually, elevated VLDL (bad triglycerides) or chylos (milky, lipid rich)
Primary cause: familial LPL deficiency, familial apo-CII deficiency
Tangier Disease
Low cholesterol, high TG, absent HDL, absent apo-A1
cholesteryl esters deposit in tonsils, lymph nodes, spleen
Correlations with CHD or CAD
Hypercholesterolemia (curvilinear, >200, related to high LDL), low HDL (<40), HDL<35 an independent risk factor, Lp(a)>30
Labeled Immunoassay
Radioactive (RIA, IRMA)
Fluorescent (FIA)
Chemiluminescent (CLA)
Enzyme (EIA)
Competitive Immunoassay
Ag* and Ag compete for binding with the Ab
Ag* concentration is constant and limited
As the Ag concentration increases, more bind with Ab and less Ag* binds, bound Ag* is always measured.
**The highest concentration of the Ag generates lowest signal and vice versa
Non-Competitive Immunoassay
Ab reagent is labeled and is always added in excess (to avoid limiting the reaction)
Concentration of the Ag is directly proportional to the bound labeled Ab up to a limit, but above that it will be prone to “hook effect”