2. erythrocytes - hemoglobin
1/59
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
60 Terms
Erythrocytes/Red Blood Cells
Small anucleated cells, with no organelles
Erythrocytes shape
Biconcave disc shape
Large surface and thin membrane ➔ O2 diffusion
Flexible membrane ➔ passage through capillaries
Erythrocytes contain
mostly hemoglobin = O2-binding protein
Role in O2 delivery to tissues and CO2 removal
Life span of erythrocytes
120 days
erythrocyte life cycle
erythrocytes form in red bone marrow
circulate in blood stream for 120 days
aged erythrocytes are phagocytized in liver + spleen
heme components of blood are recycled
heme → secreted in bile from the liver + iron stored by ferrin in the liver
membrane proteins + globin proteins → amino acids
some are used to make new RBC
Hematopoiesis
Development of all blood cell lineages from hematopoietic stem cells
primitive or definitive
Hematopoietic stem cells
self-renewing throughout the life of the host
100 billion cells produced per day!
Primitive Hematopoiesis
Transient process until embryonic w8
Cells originate from yolk sac
Definitive Hematopoiesis
During early fetal life
mostly in liver and spleen
In late fetus, the main site becomes the bone marrow
Erythropoiesis
RBC production
Erythoblasts/normoblasts → Normoblasts → Reticulocytes → mature erythrocytes
Erythoblasts/normoblasts
first recognizable red blood cell precursors
reticulocytes
Normoblasts decrease in size and lose nucleus
no longer need folic acid but need iron
Released into blood stream, circulate for 1-2 days
mature erythrocytes
Reticulocytes lose mitochondria and ribosomes
Maturation in spleen
Regulation of erythropoiesis
Regulation by blood oxygenation:
Low blood O2 ➔ tissue hypoxia
In kidney, activation of hypoxia-inducible factor 1 (HIF-1) to induce synthesis of erythropoietin (EPO)
EPO stimulates erythropoiesis (from 2.5 million cells/second (steady state) to 17 million cells/second!!!)
Increased RBCs restore normoxia and EPO levels decrease
Factors that decrease oxygenation
Low blood volume (hemorrhage)
Anemia
Low hemoglobin
Poor blood flow
Pulmonary disease
Hemoglobin
1/3 of the RBC weight
Specialized protein with heme as prosthetic group
Adult Hb (HbA)
found exclusively in RBC
Heterotetrameric hemeprotein
Heterotetrameric hemeprotein
4 polypeptide (2a and 2b) chains = globins bound by noncovalent bonds
Each chain has an a-helical structure (a has 7 helices and b has
8 helices) and a hydrophobic heme-binding pocket (prosthetic group)
Heme
Flat, planar molecule
Complex of protophorphyrin IX and Fe2+
Prosthetic group for hemoglobin, myoglobin, cytochrome
Porphyrins
cyclic molecules formed by the fusion of 4 pyrrole rings through methenyl (=CH-) bridges
Side chains define the type of prophyrin
Heme synthesis + rates in what tissues
Synthesis in liver (heme for cytochromes) and bone marrow (heme for Hb)
In liver, variable rate
In bone marrow, constant rate to match RBC regeneration
Heme synthesis subcellular location
Synthesis begins in the mitochondria and continues in the cytosol
heme synthesis steps
Formation of d-aminolevulinic acid (d-ALA) Catalyzed by ALA synthase (ALAS)
rate-limiting enzyme (DOES NOT USE ATP)
Negative regulation by heme (its own product)
2 molecules of d-ALA condense to form porphobilinogen
ALA dehydratase inhibited by Pb
4 porphobilinogens condense + side chains added
Ferrochelatase catalyzes the addition of iron ➔heme
– Ferrochelatase inhibited by Pb
Porphyrias
inherited defects in heme synthesis, resulting in accumulation and increased excretion of porphyrins
Mostly autosomal dominant inheritance
Porphyrias symptoms
Symptoms depending on the site of the mutated enzyme
Skin eruptions and wine red urine after exposure to sunlight, alcohol, or high iron intake
Red teeth (erythrodontia)
treatment of Porphyrias
Injections with hemin (Fe3+), phlebotomies, protection from light
Iron
Iron is in ferrous state (Fe2+) and can form 6 bonds:
4 with nitrogens of the rings
1 with the nitrogen of a histidine residue in globin
1 with O2
Oxidation of Fe2+ to Fe3+ (ferric) makes the molecule incapable to bind O2
where do we get iron from?
Iron is obtained from diet (most readily from heme in meats, best form)
Ferrous iron Fe2+ absorbed and oxidized to ferric iron Fe3+
Free iron is toxic ➔ transported by transferrin to bone, liver, and RBC
how is iron stored?
as ferritin (plasma indicator of amount of iron stored)
how is iron loss?
by bleeding, sweat, urine, feces, desquamation
Ideally, a balance between daily intake and output is necessary to maintain the iron stores
Globin synthesis
2 globin gene loci:
– Chr.16 – a-locus (a1, a2)
– Chr.11 – b-locus (b, d, g)
Heme is a transcriptional regulator of the globin genes
lots of availability of heme you need more globin
States of hemoglobin
Based on the level of oxygenation, Hb can exist in 2 conformations: oxygenated and deoxygenated
States of hemoglobin: T (tense) state
deoxygenated
has low affinity for O2
Strong interactions between the a and b chains to form heterodimers
Salt bridges between ab dimers constrain the movement of the chains
States of hemoglobin: R (relaxed) state
(oxygenated
has high O2 affinity
Binding of O2 to one heme results in dimer rotation with disruption of most bonds, resulting in availability of other hemes to bind O2
Erythrocyte metabolism
No mitochondria ➔ only cytoplasmic enzymes
– no damage prevention and repair proteins
– Generation of energy by glycolysis
Erythrocyte metabolism: Rapoport-Luebering shunt
used by erythrocytes use the to produce 2,3-bisphoshoglycerate
(2,3-BPG)
2,3-bisphoshoglycerate (2,3-BPG)
modulator of O2 binding
Erythrocyte metabolism: NADH role
necessary to regenerate hemoglobin from methemoglobin by NADH-cyt b5 methemoglobin reductase
Erythrocyte metabolism: NADPH role + where it is produced
produced in pentose phosphate shunt
used to maintain glutathione in reduced state (defense against damage by ROS)
uses enzyme glucose-6-phosphate dehydrogenase
Lifetime of RBC correlates directly with
glucose 6-phosphate dehydrogenase activity
Erythrocyte destruction
Erythrocytes become increasingly fragile
Proteins required to maintain plasma membrane fluidity cannot replaced
Macrophages phagocytize old RBCs (200 bil/day)
RBCs are constantly filtered through
spleen and liver
Destruction of old erythrocytes →
release of hemoglobin
Heme:
• Iron → iron cycle
• Biliverdin → bile pigments
Globin → AA metabolism
Heme degradation
Heme is oxidized and linearized to biliverdin and CO by heme oxygenases and NADPH
Fe2+ is released and returned to iron stores
Biliverdin (green) is reduced to bilirubin (red)
Bilirubin is transported to the liver bound to albumin
In the hepatocytes, bilirubin (UCB) is conjugated twice with glucuronic acid
Bilirubin diglucuronide (CB)is actively secreted in the bile
Iron cycle
Iron released from the macrophages is largely Fe2+
Multicopper ferroxidase ceruloplasmin converts Fe2+ to Fe3+
Fe3+ transported bound to transferrin and delivered to most cells via a cell surface transferrin receptor 1(TfR1)
The receptor-transferrin complex is then internalized by receptor-mediated endocytosis and bound iron is released in the cytosol and transferrin is recycled
Sickle cell disease (AR)
Common in African populations and descendants (heterozygous advantage)
Point mutation (GAG→ GTG) results in glutamate being replaced by valine in the b-chain
HbS formed by large, linear polymers (needle-like) causing the erythrocytes to become sickle-shaped under conditions of:
■ Low oxygenation
■ Low pH
■ Dehydration
Sickling
at first reversible
Cumulative damage to RBC membrane with repeated sickling episodes leads to permanently sickled RBCs even under normal physiological conditions
Fragile RBCs ➔ intravascular hemolysis
RBC aggregate in capillaries ➔ microvascular occlusions
Sickling specific to microvascular beds with slow transit times (spleen, bone marrow, inflamed vascular beds)
Sickle cells anemia (homozygotes)
85-95% HbS and rest HbF
Severity of disease increases with the increase of the proportion of HbS in deoxy (t) state
Blood smear: irreversibly sickled RBCs, reticulocytosis
Sickle cells anemia (homozygotes) manifestations
Severe hemolytic anemia, vasocclusive crises (bone, lungs, spleen, CNS), multiorgan infarctions
Sickle cells anemia (homozygotes) manifestations in spleen
Sequestration crises ➔ massive pooling of sickle RBCs in the spleen ➔ splenic enlargement
Repeated splenic infarctions ➔ fibrosis ➔ progressive atrophy
Oral aspects of SCA
Mandibular osteomyelitis and bone marrow hyperplasia ➔ enlarged maxilla and overbite
Microvascular occlusions ➔ painful infarcts ➔ radiolucency followed by
osteosclerosis
Coarse trabecular bone pattern
Cranial X-ray: hair-on-end appearance
Smooth reddish tongue with atrophied papillae
Sickle cell trait (heterozygotes)
55-75% HbA, 25-45% HbS
Generally asymptomatic
Sickling can be triggered only by severe stress, high altitude, dehydration, or strenuous physical exercise
Normal RBC morphology
a-thallasemias
Gene deletions ➔ decreased/absent a-globin
Phenotype varies from mild to severe depending on the number of gene copies affected
autosomal recessive
types of alpha thallasemias
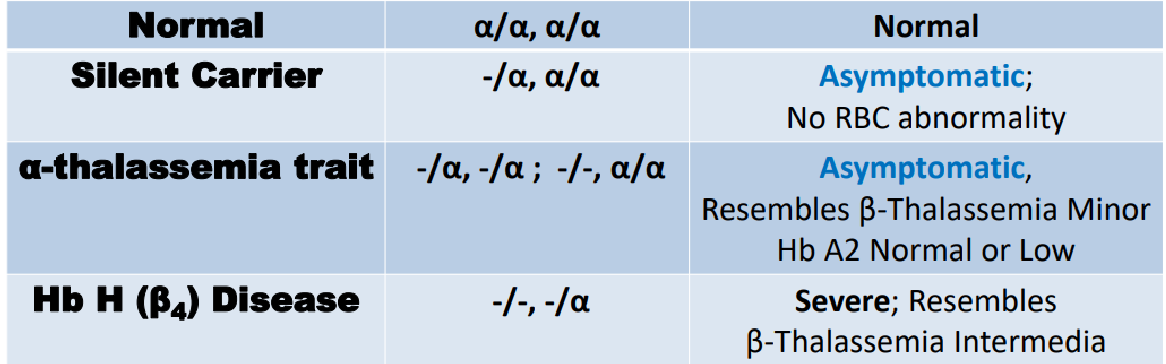
alpha thallasemias Symptoms due to:
Insufficient hemoglobin (all Hb types affected)
Excess unpaired globin chains polymerize ➔ forms of Hb with inadequate affinity for O2 ➔ tissue hypoxia
Excess g-chains in fetal period ➔ g4 (Hb Barts) replace HbF
Incompatible with life
Excess b-chains in children and adults ➔ b4 (HbH) replace HbA
HbH prone to oxidation and precipitation ➔ intracellular inclusions
Severe anemia, enlarged spleen, bone deformities
b-thallasemias
Point mutations ➔ decreased/absent synthesis of b- globin
autosomal recessive
types of b-thallasemias
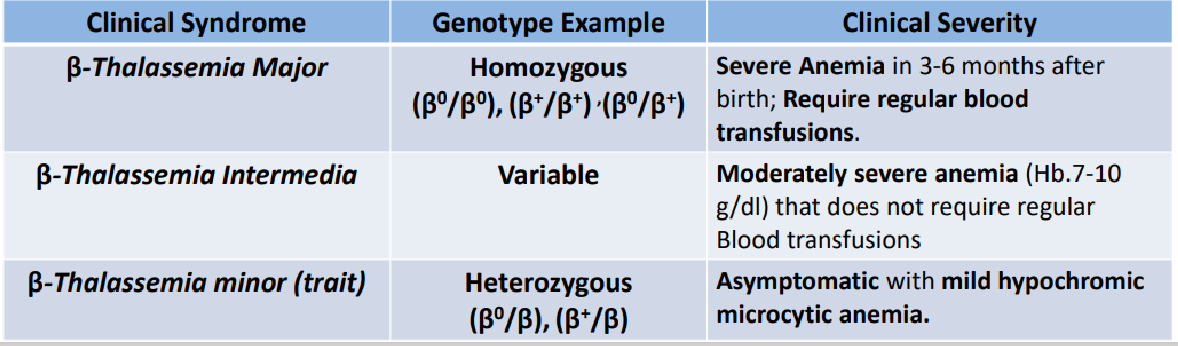
b-thallasemia major
Reduced b-globin synthesis → reduced HbA ➔ severe anemia
Aggregation of excess a-globin
Cell damage ➔ extravascular hemolysis ➔ jaundice
Abnormal erythroid progenitors ➔ ineffective erythropoiesis ➔anemia
Excess erythropoietin ➔ extramedullary hemopoiesis (hepatomegaly, splenomegaly)
Severe marrow hyperplasia ➔ skeletal abnormalities
Repeated blood transfusions ➔ systemic iron overload (secondary hemochromatosis)
Bone Abnormalities in b-thallasemia major
Abnormal facial features (large cheekbones, depressed nasal bridge, protruding maxilla)
Hair-on-end appearance on cranial X-rays
Osteoporosis → fractures
Delayed puberty, endocrine disturbances (due to anemia and iron overload)
Higher incidence of chronic hepatitis B&C due to blood transfusions
Peripheral blood smear: severe hypochromia microcytosis, numerous nucleated RBCs
b-thallasemia minor trait
Usually asymptomatic
Mild anemia (1-2 g/dl below normal)
Peripheral blood smear: hypochromia, microcytosis, nucleated RBC, poikilocytosis