L3: Applying massive parallel sequencing (Pt2)
1/51
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
52 Terms
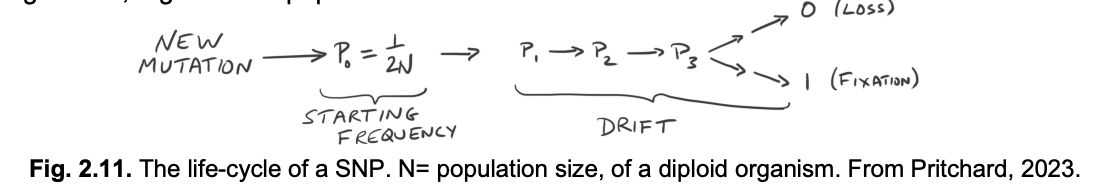
Evolution and SNPs: SNP life cycles
SNPs are born by mutation and may drift in frequency of face selection over their evolutionary life cycle
General tendancies:
Neutral SNPs (most SNPs)→ drift up or down in frequency
eventually facing loss or fixation
Deleterious SNPs→ usually remain rare
until lost by selection
Advantageous SNPs→ usually increase in frequency
→ towards fixation
Note: they are all subject to random fluctuations which can be signficiant e.g in small populations
N here is the population size (se E and B notes for how this equation is derived
Gene divergence
sequence divergence between species from a common ancestor
which gives rise to ‘orthologs’
(as opposed to paralogs→ form by gene duplication within a species)
Divergence rate reflects…
a molecular clock:
Allows estimations of:
divergence time
phylogenetic relationships
Clock speed can vary among
proteins
species
THERFORE→ can result in misleading trees→ if interpretation is too naïve
How to detect positively selected alleles that drive evolutionary change?
Some causative changes should show traces of positive selection:
higher rate of change or polymorphism for ‘non-synonymous’ positions that cause amino acid changes
compared to ‘silent’ nucleotide changes→ e.g 3rd codon positions
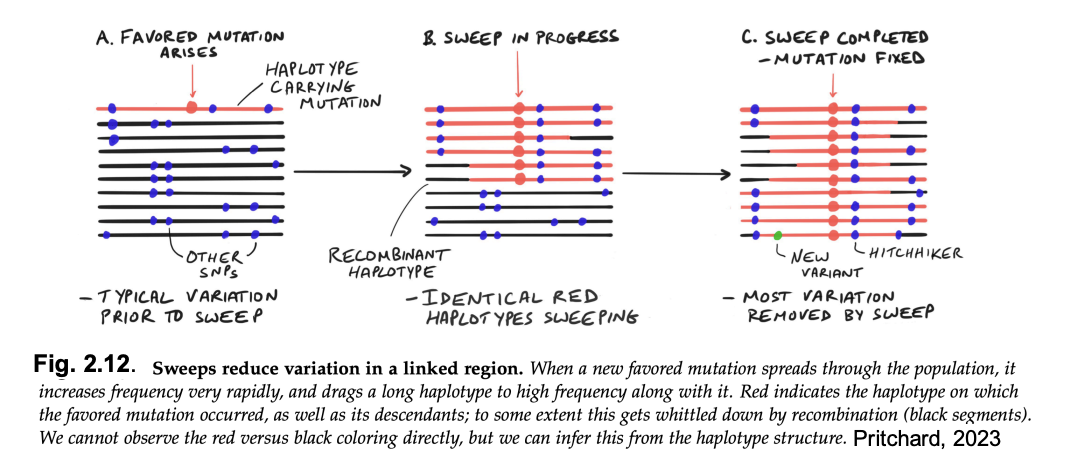
selective sweep→ new mutation is selected for and linked polymorphisms are swept (or hitchhike) along with it
see examples below
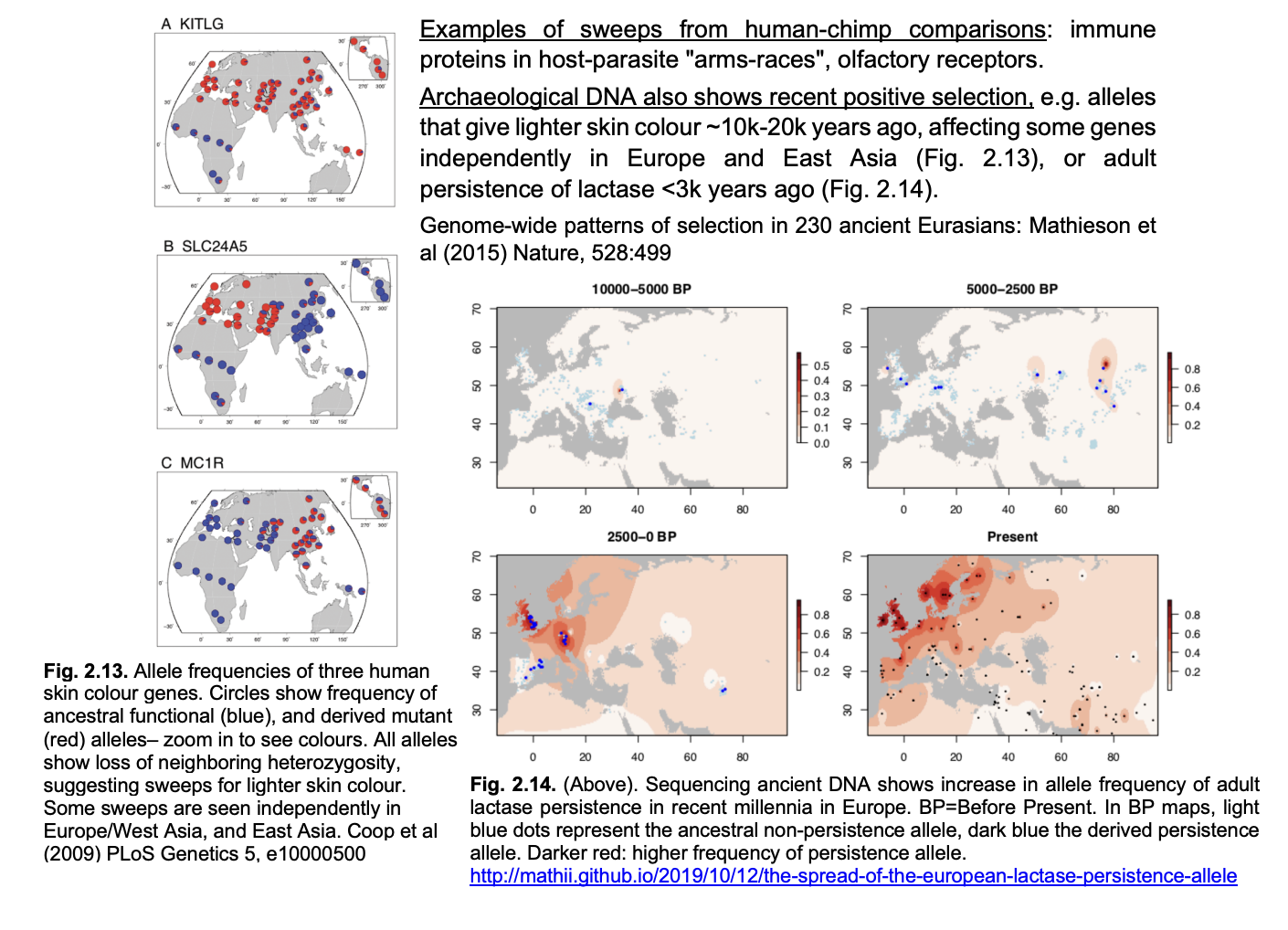
Examples of sweeps from human-chimp comparisons
immune proteins in host-parasite ‘arms-races’
olfactory receptors

Other evidence: archaeological DNA shows recent positive selection
e.g 1→ alleles that give lighter skin colour→ 10k-20k years ago
affecting some genes independently in Europe and East Asia
e.g 2→ adult persistence of lactase→ <3k years ago
Other sweeps in human populations
Immune loci→ specific disease resistance
Altitude→ genes controlled by HIF (hypoxia-nduced factor)
selected haplotype in Tibetans appears to originate from Denisovan hominins
Arcitic diet: decreased fatty acid import into mitochondria

Nucleotide variation: sum up of SNPs
SNP are reflection of out individuality and a snap shot of evolution in action
Common SNPs→ useful tool for GWAS and mapping inheritance of common traits
BUT: account for only a fraction of hertibability
Rare alleles→ from large banks of exome and genome sequences
coupled with phenotpyic data
→ VALUABLE source of genetic data on human traits and diseases
Selected differences between different human populations→ informative about the genetics of human physiology
HOWEVER: there are still large gaps in resources in non-white populations
Structural variants in genomes and how they evolve:
Copy number variants:
these are duplications and deletions of DNA
How do copy variants arise
Polypoidisation
Unequal Crossovers
Polyploidisation
duplicates every gene in the genome→ whole genome duplication (WGD)
E.g in yeast→ had such duplication and THEN loss of 90% of duplicated genes
E.g→ evidedence for similar eveents in telost fish
Unequal crossovers
give rise to tandem gene families
perhaps initially between two copies of repetitive sequence
THEN
between copies of the duplicated genes
Allows for→ frequent changes in copy number during evolution
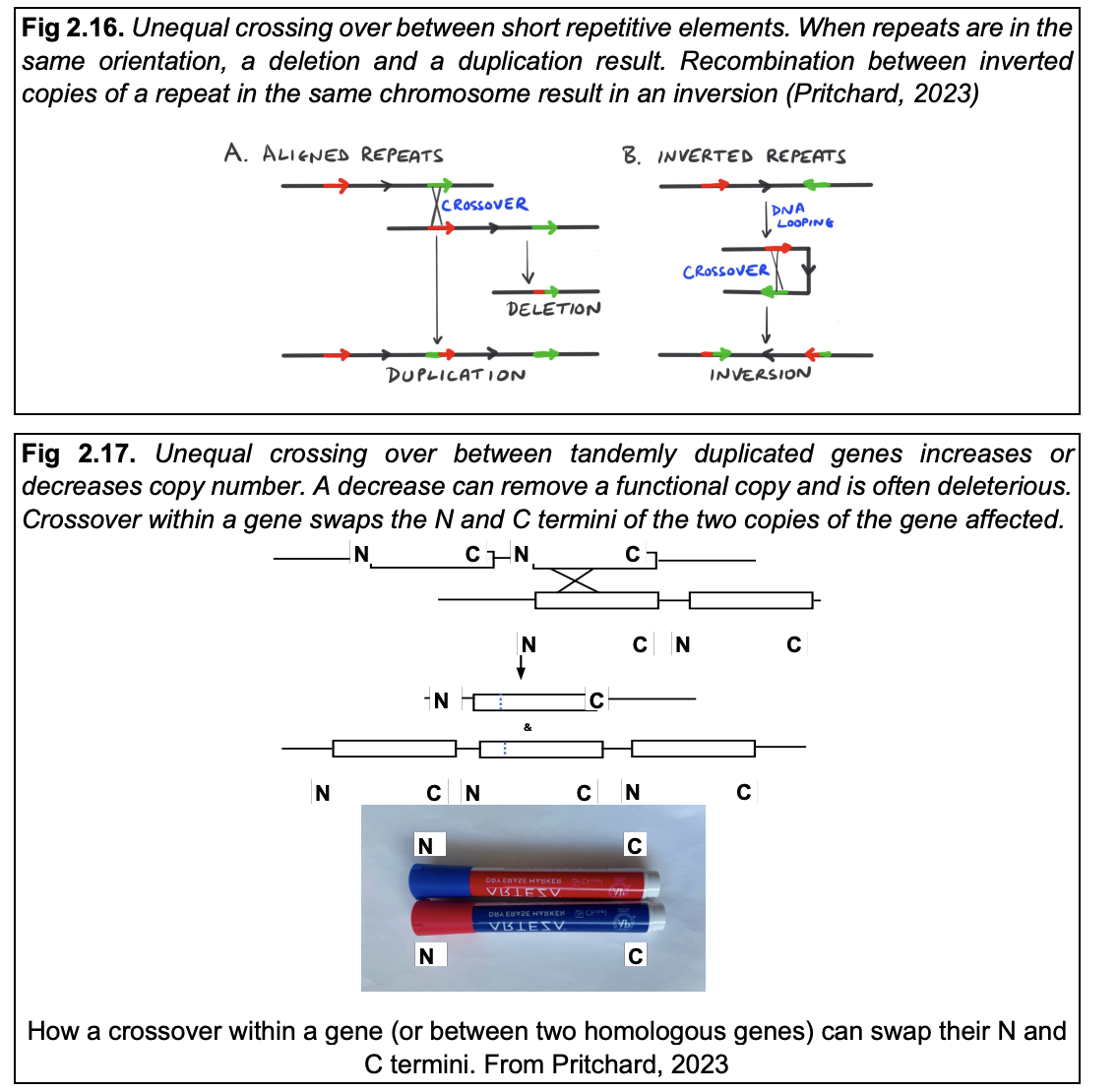
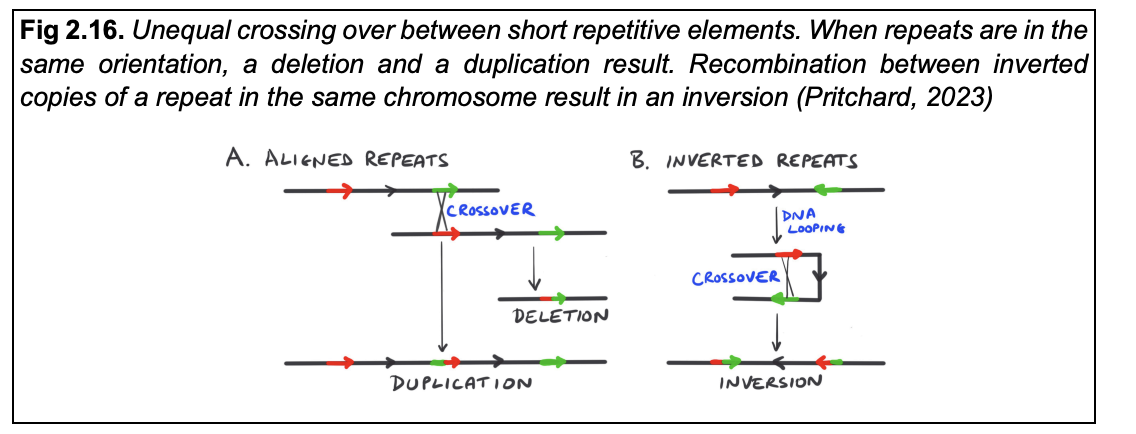
Unequal crossing over→ between short repetitive elements
when repeats are in the same orientation→ a deletion and a duplication result
recombination between inverted copies of a repeat in the same chromosome result in→ an inversion
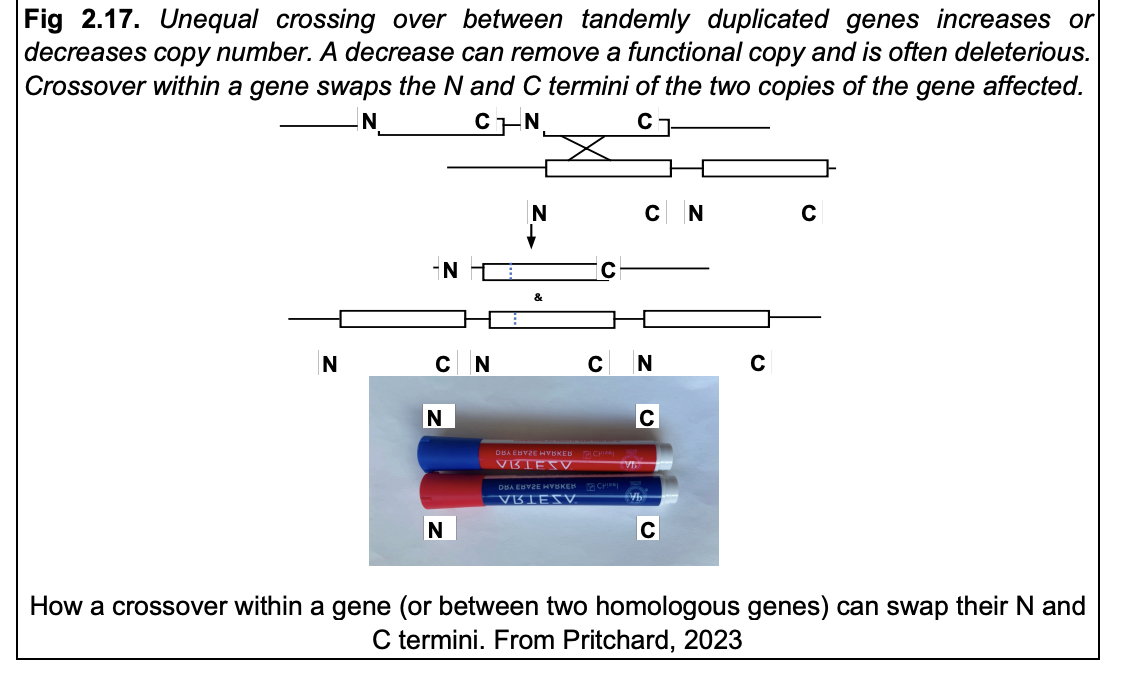
Unequal crossing over→ between tandemly duplicated genes
increases or decreases copy number
A decrease can:
remove a functional copy
is often deleterious
Crossover within a gene:
swaps the N and C termini of the two copies of the gene affected
Detecting counting and characterising strucutural variants: originally identified from phenotypes
Although given the frequency of indels and CNVs→ much probably without major phenotypes there are still some CNVs which have phenotypic consequences
Thalassemias→ unequal crossovers in globin genes)
Colourblindness→ unequal crossovers in red and green opsin genes
themselves originated only recetly as a duplication in primates
Prader-Willi syndrome→ duplication of chromosome 15 that cause intellectual disability
The more carefully we look for structural variants, the more we have found

Other ways of selecting structural changes→ sequencing a few individuals
detected hundreds of structural changes relative to the ‘reference’ sequence
90% insertions or deletions (indels)
Ways to do this:
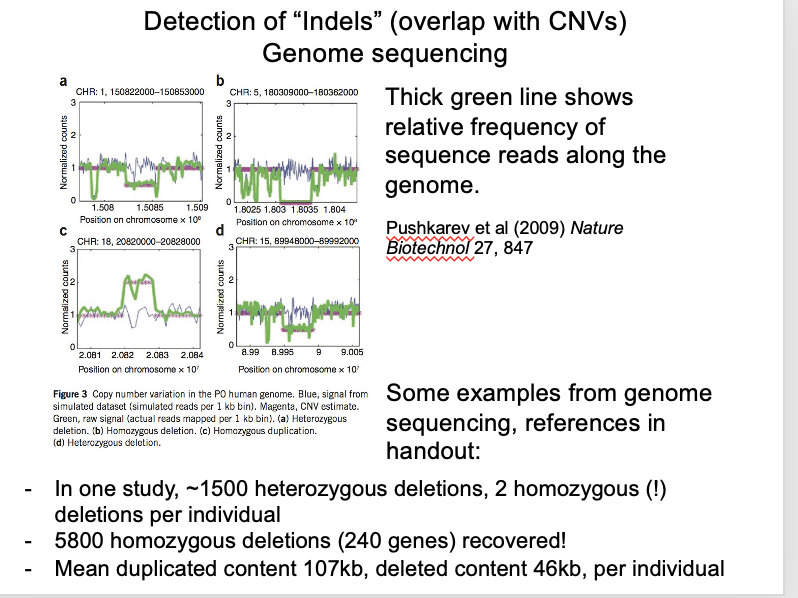
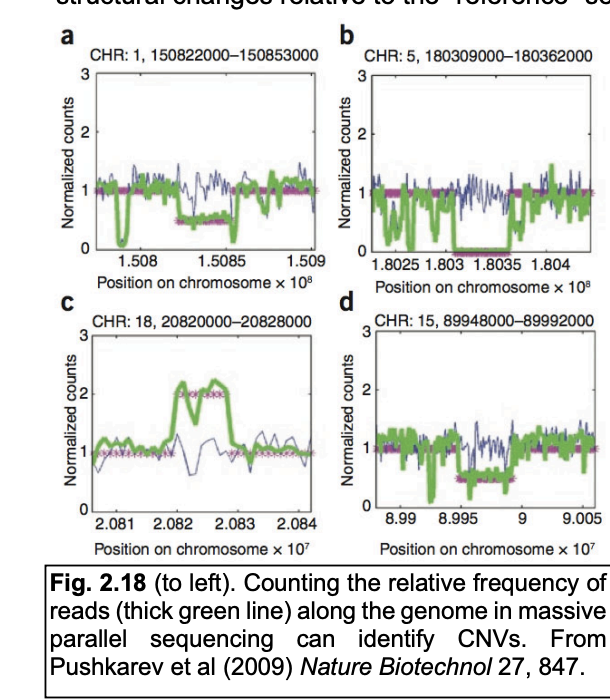
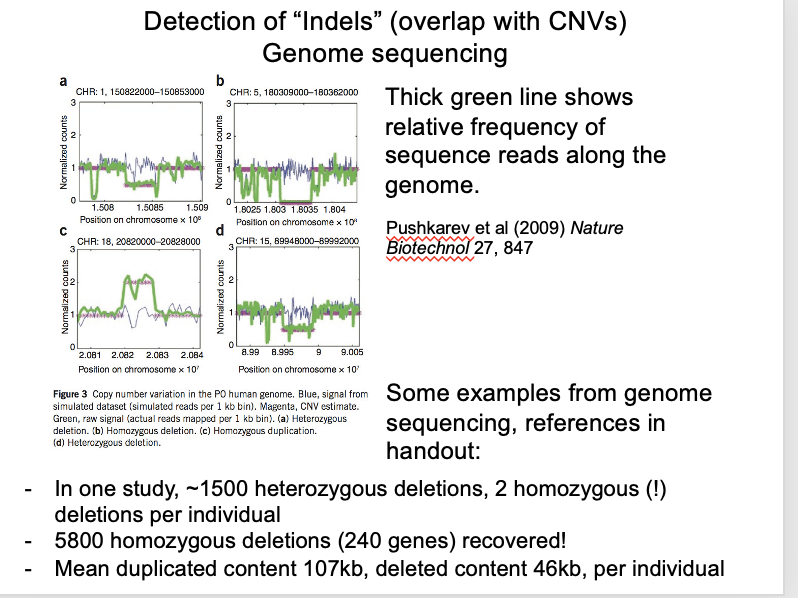
Local read frequency (illumina) in short-read genome sequencing
Long-read sequencing (nanopore)
Local read frequency (illumina) in short-read genome sequencing
long read frequency improves resolution and detects more variants
e.g in 2500 whole human genomes
each individual showed 1500 heterozygous and 2 homozygous deletions
Local read frequency (illumina) in short-read genome sequencing: OVERALL what has been found
5800 homozygous deletions in this study covered 240 genes
dispensable for human life
→ Time will tell how these individuals manage without them
Graph:
Blue line→ control
Green to the bottom→ homozygous
Green to halfway to the bottom→ heterozygous
Long read sequencing (Nanopore)
Identified even more variation:
e.g in 3622 Icelanders, >22k structurual variatns per individual
(13k insertions, 9k deletions)
A few are associated with phenotypes→ e.g low LDL, height)
Most aren’t
OVERALL: the bottom line
We have a lot of structural variation:
affecting much more of our genome than SNPs
Some of it can cause genetic disease
BUT
A surprising amount DOES NOT
Evolutionary consequences of this
increased gene number
Increased gene number: fate
Homologous genes formed by duplication in the same species are PARALOGOUS GENES
Have two (or three?) fates:
Redundancy of function→ allows one to mutate to non-functional (pseudogenes) over several Myr
New/ more specialised functions→ increase developmental and physiological complexity→ multigene families
Increased gene dosage→ e.g rRNA, histones
Duplications are a source of developmental and physiological complexity
e.g gobin and Hox genes
Duplication of protein domains→ new protein architectures with combinations of domains
Truly new domains appear occasionally (but hard to verify)

About 1000 types of domains…
give rise to diversity of proteins we see today by
duplication and divergence
VNTRs
Variable number tandem repeats
→ short repeats with higher copy number
Types:
satellite sequences
Minisatellites
microsatellites

Satellite sequences, features
WHAT: tandem repeats, generally heterochromatic, condensed
WHERE: most in centromeric regions
some in telomeric regions
SIZE: vary from few bp→ several hundred
COPY NUMBERS: 104→ 106
HOW MUCH OF GENOME: 10-40% of the genome
humans→ 10%

Why is there so much satellite DNA?
Questions to ask:
does it have a function?
Selfish?→ replicates at expense of the rest of the genome or the organism
Tolerated?
Junk? no function
What Drosophila chromosomes with large satellite deletions suggest:
not much (if any) is needed
Mammalian centromeres evidence?
long repeat arrays (unlike yeast)
BUT→ if one array is deleted→ an array with different sequence can substitute as a new centromere
Mini/micro satellites compared to satellites
more dispersed through the genome
Minisatellites
COPY NUMBER: 3-100 copies
LENGTH: 30bp
WHERE: 103 locations per genome
Microsatellites
COPY NUMBER: 10-50 copies
LENGTH: 2-6 bp
WHERE/HOW MANY IN MAMMAL GENOME: only every few kb→ >105 per mammal genome
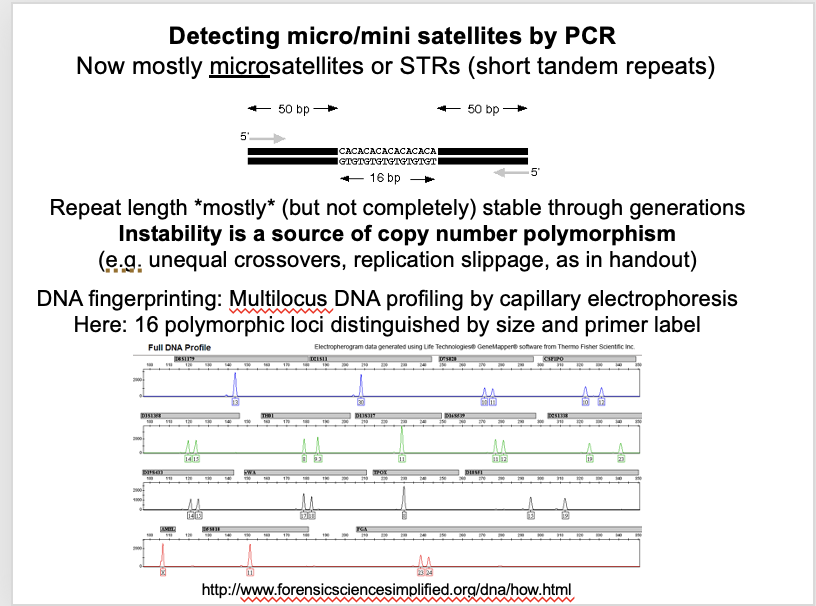
PCR analysis of these shows: (and applications)
REPEAT NUMBER:High degrees of polymorphism in repeat number→ allows DNA fingerprinting
INSTABILITY OF NO.: High rate of instability in repeat number→ 0.1-1% per locus per generation
a bias towards increased repeat number in succeeding generations
DIVERGENCE: A small degree of sequence divergence between repeats
for minisatellites
Expected→ since homogenisation of an array is not instantaneous
as the microsttelites do not really change too much over generations→ can be used for DNA fingerprinting→ because it forms different polymorphisms
Mechanisms of tandem repeat dynamics
Unequal crossing over
Replication slippage
Selection
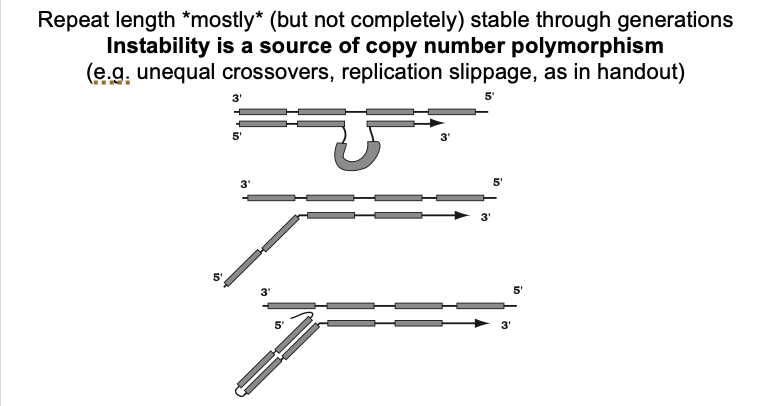
Unequal crossing over
no bias towards gain or loss
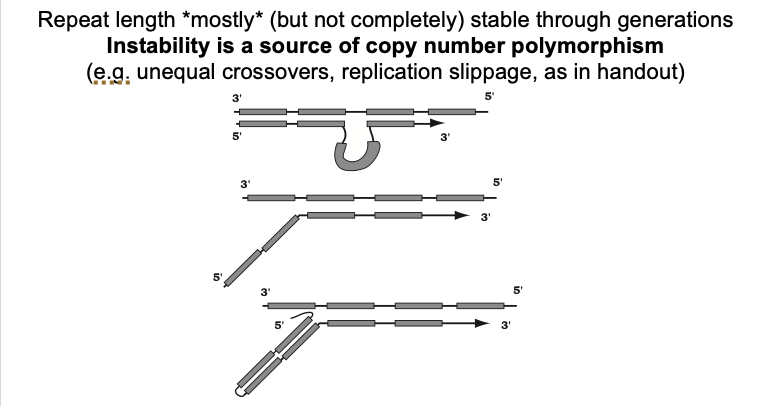
replication slippage
Could give either gain or loss of repeats
In Vivo→ there seems to be a bias towards gain
WHY: from stable 5’ ‘flaps’ hard to remove from okazaki fragments
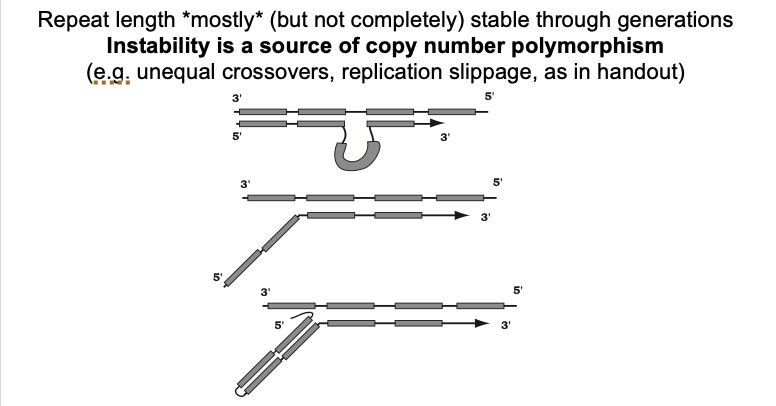
Selection
most eukaryotic genomes can tolerate long repeat arrays
BUT
at some length→ there must be selection against long arrays
this will reduce average repeat number
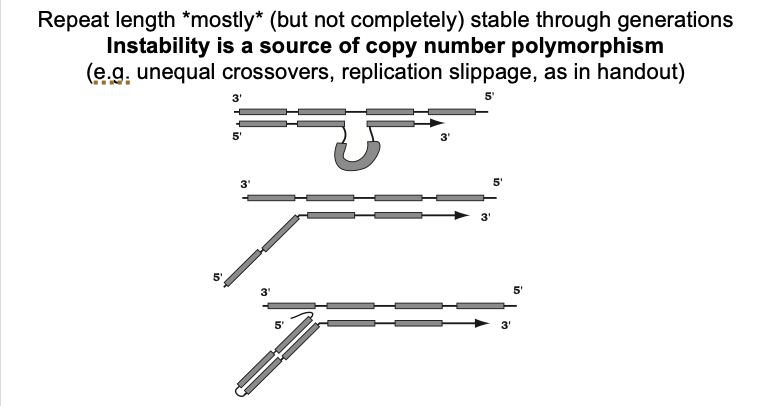
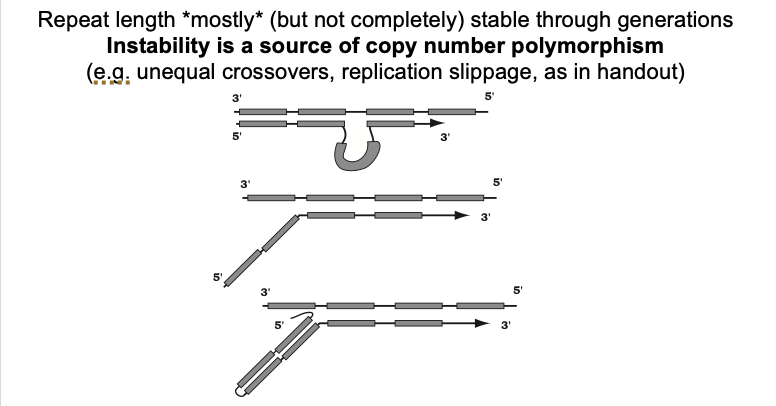
What this diagram shows: Replication slippage
Daughter strand looping out→ due to mispairing
Unpaired 5’ end of a daughter strand in an okaski fragment
a stable G rich non-Watson-Crick secondary structure
more stable than pairing of the entire okazaki fragment
fragment to the template
i.e→ in yeast→ when there is a mutation in the FLAP-endonuclease
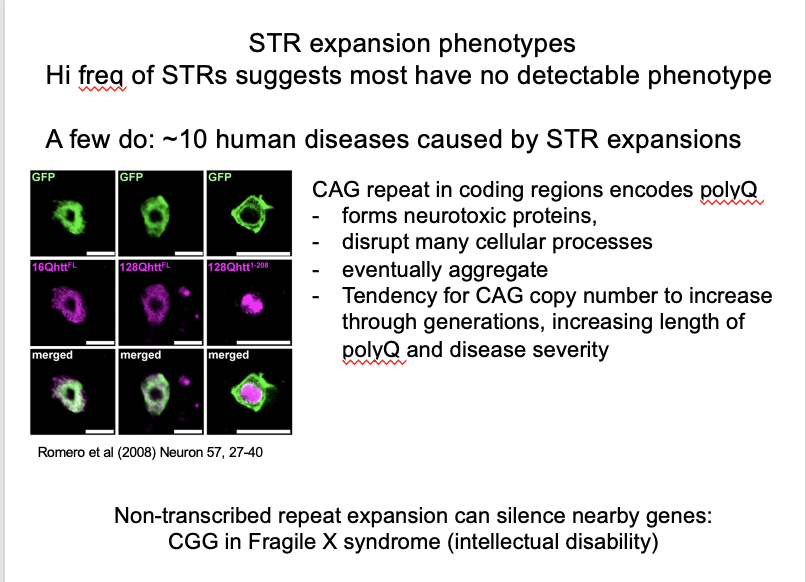
Deleterious effects of repeat expansion→ pathogenic microsatellites example:
Huntington’s Disease (HD)
What is HD gene
has a CAG repeat
encodes a polyQ stretch in the protein
prone to aggregate as polyQ length expands
forms neurotoxic proteins
disrupt many cellular processes
aggregate
toxic to cells
note: polyQ is poly-glutamine
HD gene in most humans vs affected
Normal→ 10-35 CAG copies of CAG
Affected→ >40
What does more repeats mean
more severe degeneration
Repeat severity over generations
increase through generations
until onset is early and early death leads to dying out of the expansion
Why can it take decade for HD to emerge, if expanded polyQ is toxic to cells?
Evidence from: single cell sequencing of neurons from fresh postmortem brain:
somatic repeat expansion:
CAG repeat length grows slowly with age
but only becomes toxic above repeat numbers of 150 AND critical for neuronal survival above 350
Single cell sequencing shows:
distribution of repeat lengths in patient neuronal populations
even as high as 500 copies
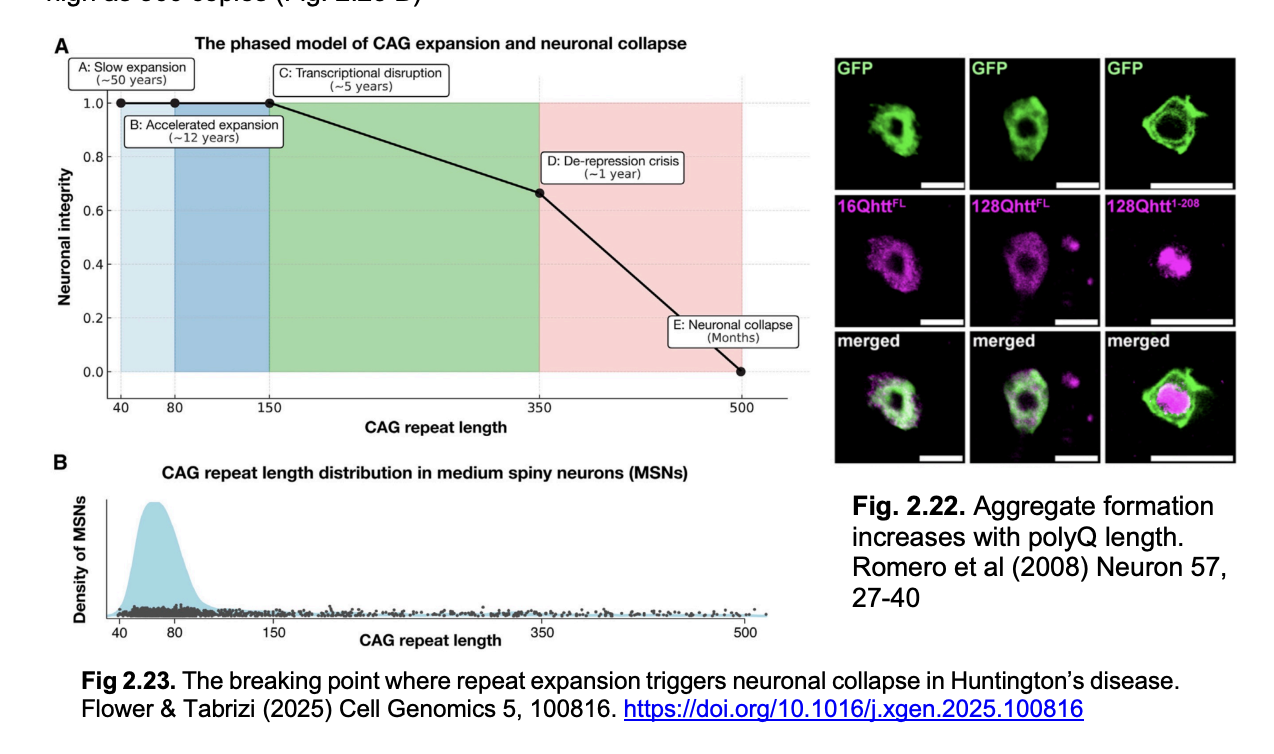
Overview of CNV/indel and repeat behaviour
copy number variants are polymorphic→ a few have phenotypes
Repetitive DNA is dynamic in populations→ with a balance between expansion and loss
Variation among individuals a snapshot of evolution in action→ as for SNPs
Human genetic Variation and Racism
Human genetic variation→ not about racisim
only about understanding out genomes, their consequences for biology and human evolution
Racism→ social value judgment→ BUT gets dressed up as smattering of biology
How can racism influence how human genetics is studied (example)
Low representation→ of fenetic diversity among the human ‘pangenome’ (although improving)
Analysing human genetic variation:
without informed involvement,
and prospect of benefits to
→ the diversity of groups participating
Eugenics
that improving the human gene pool outranks the worth of certain human individuals
usually not from the same ethnic, society or white racial group the eugenics is from

Consequences of eugenics
discriminatory social policy
through forced sterilsation
involuntary euthanasia→ mutder
genocide
Eugenics still active
2 child benefits policy
views of the current US administration on autism or infectious disease susceptibility
white supremacy
IQ
IQ
has a genetic component like all human traits
BUT
to measure pparant IQ racial differences and conclude this is genetic is gross extrapolation
Example of IQ
Richard Lynn:
Irish low average IQ
seen as an impediment to advancement of the Irish population
BUT
mysteriously climbed by 10% points in a generation
Extremely dangerous consequences
‘certain groups must be left to fend for themselves because nothing can be done for them and the rest of us matter more’
Why should IQ be associated with skin colour any more than with hair or eye colour?
Overall, the more you understand genome variation, its cuases and consequences
the more obvious will be the flaws in ‘scientific’ racism and eugenics