W11L2 - lysosomes
1/39
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
40 Terms
What are lysosomes?
Membrane enclosed organelles containing 40 hydrolytic enzymes
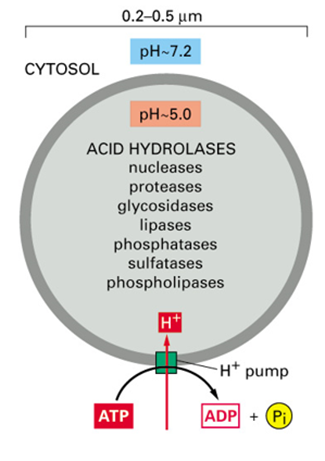
What can lysosomes degrade?
proteins, nucleic acids, sugars, lipids and pump products to cytoplasm
Can degrade defunct intracellular organelles or macromolecules and particles taken in by endocytosis
What pH are lysosomes maximal activity?
5
How is low pH of lysosomes maintained?
Vacuolar v ATPase
What does the low pH help to do?
denature proteins - better accessibility for proteases
What protects lysosomal membrane proteins from proteases?
They are highly glycosylated
Where are lysosomal enzymes synthesised and then where are they sorted to?
• Lysosomal enzymes synthesised at ER and transported to Golgi
• At TGN sorted into clathrin-coated vesicles for transport to late endosomes, that subsequently fuse with lysosomes
How does this sorting occur?
Lysosomal enzymes are tagged with mannose-6-phosphate (M6P) on N-linked oligosaccharide as they pass through lumen of cis golgi network
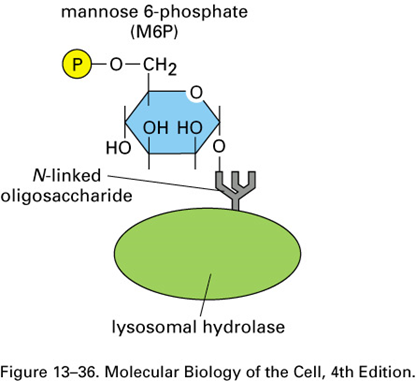
ER - How does the golgi know which glycoproteins to tag?
This requires specific recognition of the hydrolases by the Golgi enzymes responsible for adding M6P.
Because all glycoproteins leave the ER with identical N-linked oligosaccharide chains, the signal for adding the M6P units to oligosaccharides must reside somewhere in the polypeptide chain of each hydrolase.
Genetic engineering experiments have revealed that the recognition signal is a cluster of neighboring amino acids on each protein’s surface, known as a signal patch
Because most lysosomal hydrolases contain multiple oligosaccharides, they acquire many M6P groups, providing a high-affinity signal for the M6P receptor
How is M6P added in the golgi apparatus?
o Addition of phospho-GlcNAc to mannose by GlcNAc phosphotransferase
o removal of GlcNAc to leave mannose 6-phosphate tag
What happens when the enzyme tagged with M6P gets to the TGN?
When the enzyme with M6P tag gets to TGN, it binds to clathrin adaptor proteins – AP1 and GGA, to mediate the incorporation of the hydrolyase into a clathrin coated vesicle
bud off the TGN and take cargo to an early endosome
In the early endosome, the low pH results in the dissociation of hydrolyase from M6P receptor (pH in early endosome = 6)
Receptor is recycled back to TGN using sorting nexin proteins
Phosphate gets removed from hydrolase
Endosome will mature
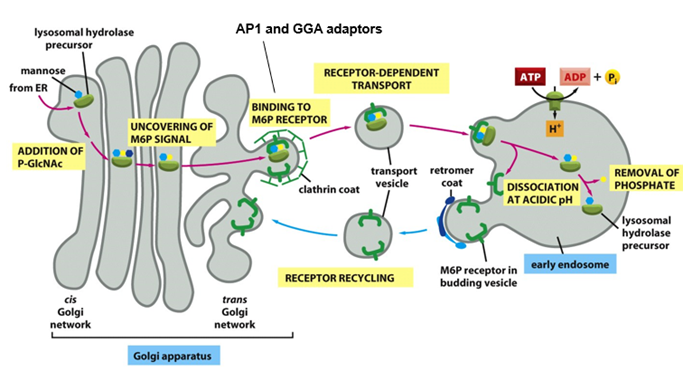
ER - At what pH does the M6P receptor bind to M6P?
pH 6.5-6.7 in the TGN lumen
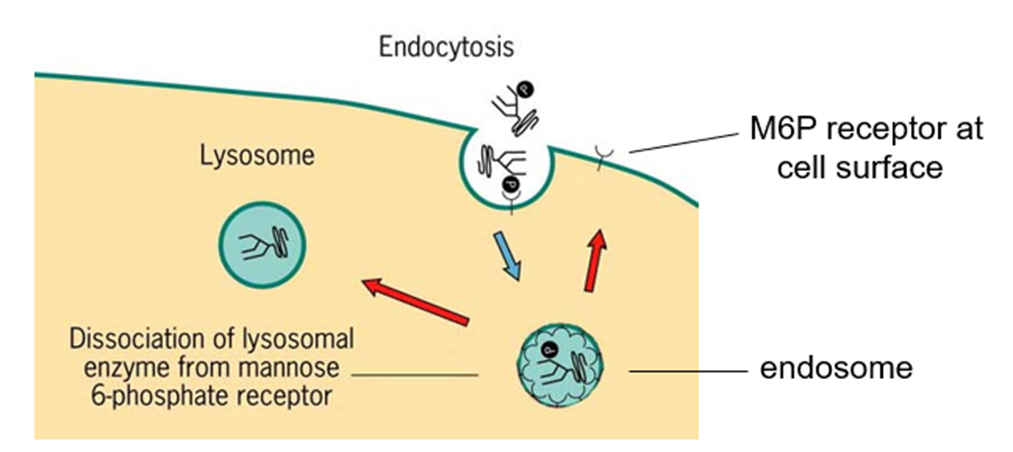
How can some M6P tagged enzymes escape the normal packing process?
They are secreted into the extracellular fluid by constitutive secretory pathway
How are lysosomal enzymes retrieved from extracellular fluid?
• Most cells in the body also have M6P receptor on PM
• Some M6P receptors also cycle to PM => bind enzymes and return them by receptor-mediated endocytosis to lysosomes via early and late endosomes
• Lysosomal enzymes require an acidic environment to work so do little harm in extracellular fluid (pH7.4)
What are lysosomal storage diseases?
• Genetic disorders
• Accumulation of undigested substrates - pathological effects
• Loss of hydrolytic enzymes from lysosomes
What is the most severe lysosomal storage disease?
Inclusion (I) cell disease
What happens in I cell disease?
o ALL hydrolytic enzymes missing from lysosomes due to a recessive gene defect
o Secreted into blood using ‘default pathway’
o Due to mutation in gene for GlcNAc phosphotransferase => no M6P tag added to lysosomal hydrolases
What cell types are not affected by I cell disease?
Not all cell types affected (e.g. hepatocytes) - implying that there is another pathway for directing hydrolases to lysosomes that is used by some cell types
Lysosomal membrane proteins not affected => M6P-independent pathway also exists
What are examples of other lysosomal storage diseases?
Gauchers
Deficient of glucocerebrosidase
Hurlers (ER)
the enzyme required for the breakdown of certain types of glycosaminoglycan chains is defective or missing
How can Gauchers be treated?
enzyme replacement therapy
• Enzyme back in bloodstream so M6P receptors on cell surface will allow it to be taken up
• Super expensive
• Continual injection of enzyme
What is currently undergoing clinical trials to treat lysosomal storage diseases?
Gene therapy to reexpress enzymes
What are the different pathways to lysosomal degradation?
• Phagocytosis - uptake of large particles by specialised cells (e.g. macrophages)
• Endocytosis - specific uptake of small molecules
• Macropinocytosis - non-selective uptake of fluid and molecules
• Autophagy - removal of damaged organelles and protein aggregates
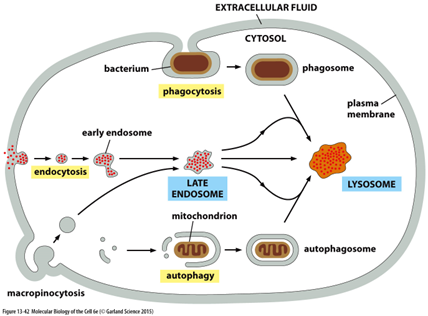
Describe lysosomes
heterogeneous
• Diverse morphology reflects wide variety of digestive functions
• Common feature = high content of hydrolytic enzymes
What is the function of autophagy?
Removal of organelles
Also for removal of protein aggregates, intracellular bacteria during infection, regions of cytoplasm during starvation
What is the structure of an autophagosome?
Double membrane
ER - Is autophagy selective or non-selective?
Autophagy can be either nonselective or selective. In nonselective autophagy, a bulk portion of cytoplasm is sequestered in autophagosomes. In selective autophagy, autophagosomes tightly enclose specific cargo and mostly exclude the surrounding cytosol
What do defects in autophagy cause?
Infection, neurodegeneration and cancer
For neurodegeneration, protein aggregates build up (eg Tau in Alzheimers) – autophagy can clear these
How does autophagy work (some ER)?
Begins when a phosphoinositide lipid kinase complex (ATG1) locally produces PI3P to mark potential membrane site for recruitment of autophagy related factors
These factors catalyze the covalent attachment of the membrane lipid phosphatidylethanolamine to a ubiquitin-like protein called ATG8
The ATG8-marked vesicles undergo homotypic fusion with each other and heterotypic fusion with vesicles containing ATG9
This results in expansion of a membrane whose identity is provided by ATG8 and other autophagy-related proteins to form autophagosome
The outer membrane of this newly formed autophagosome then fuses with the lysosome in a SNARE-mediated process.
The inner membrane and its enclosed cargo are released into the lysosome where they are degraded by the acid hydrolases
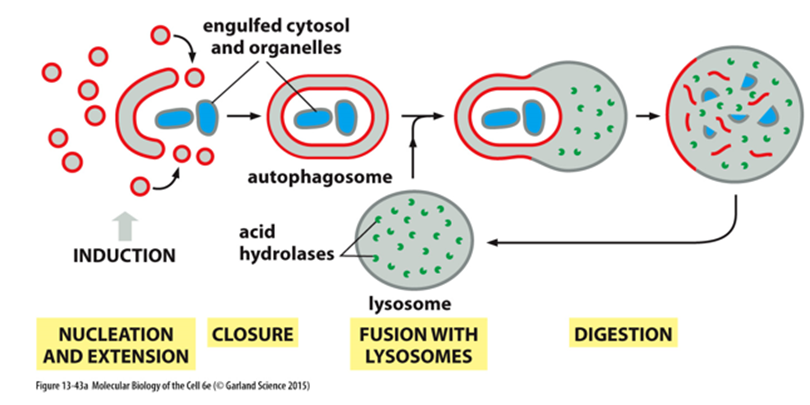
What is the structure of the autophagosome?
For reasons that are not known, the growing membrane structure formed by vesicle fusion is not spherical. Instead, it is a flattened disc (similar to a Golgi apparatus cisterna) that curls into a cup-shaped structure. Fusion of the lips of this cup encloses the contents inside a compartment that is now surrounded by two membranes
What are secretory lysosomes?
Specialised lysosomes found in some cell types that fuse with the PM to release contents
o e.g.melanocytes, cytotoxic T cells, sperm cells
What do melanocytes and sperm cells release?
Melanocytes = melanin, sperm cells = enzymes to degrade egg,
What do cytotoxic T cells release?
Cytotoxic T cells contain lytic granules
o Fusion with PM releases perforin and granzyme B
o Perforin forms pores in target cell- granzyme B enters and induces apoptosis
Where are vacuoles found?
Plants and fungi
How much cell volume do vacuoles occupy?
Over 30%
What is the structure of vacuoles?
• Contain hydrolytic enzymes – related to lysosomes
• Low pH maintained by a v-type ATPase
What is the function of vacuoles?
degradation and storage of nutrients
regulate turgor and cell size
What is the turgor pressure of vacuoles?
5-20 atmospheres
How do vacuoles prevent wilting?
Turgor pressure (5-20 Atmospheres) pushes outward against rigid cell wall to prevent wilting
Controlled by varying osmotic pressure of vacuole and cytoplasm
Achieved by varying synthesis/breakdown rates of polymers and transport of metabolites across vacuole and plasma membrane
How does turgor drive cell enlargement?
A plant cell can achieve a large increase in volume without increasing the volume of the cytosol.
Localized weakening of the cell wall orients a turgor-driven cell enlargement that accompanies the uptake of water into an expanding vacuole.
The cytosol is eventually confined to a thin peripheral layer, which is connected to the nuclear region by strands of cytosol stabilized by bundles of actin filaments
Key points
