CHEM 4680 / Topic 1b: Coordination Chemistry and Crystal Field Theory
1/33
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
34 Terms
What is the coordination number of a complex usually equal to?
Number of lone pairs donated to the central atom or ion.
For d-block metals, coordination numbers depend on three criteria. What do they depend on? Of these two criteria, what does coordination geometry depend on?
Coordination numbers depend on:
Size of central atom, which forms a proportional relationship with the coordination number.
Steric effects, i.e. size of ligands, which forms an inversely proportional relationship with the coordination number.
Electronic effects between central atom and ligands, explained by CFT.
Of these, coordination geometry depends on:
Steric effects. The lowest-in-energy geometry is most favoured with least inter-ligand repulsion.
Electronic effects. The lowest-in-energy positions of electrons is most favoured for energy stabilization.
With a CN = x, what are its possible coordination geometries? Which of these are sterically preferred, as per VSEPR?
x = 2 (Hint: There are two).
x = 3 (Hint: There are three).
x = 4 (Hint: There are three).
x = 5 (Hint: There are two).
x = 6 (Hint: There are two).
x = 7 (Hint: Only one).
Linear, bent.
Trigonal planar, trigonal pyramidal, T-shaped.
Tetrahedral, square planar, seesaw.
Trigonal bipyramidal, square pyramidal.
Octahedral, trigonal prismatic.
Pentagonal bypyramidal.
If we wanted to ensure idealized symmetric geometry, what can we do, ligand-wise?
Ensure all ligands must be the same.
There are two types of complexes based on identity of ligands. What are they? Define each.
Homoleptic complexes: Complexes in which all ligands are the same.
Heteroleptic complexes: Complexes in which all ligands aren’t the same.
Defining more terminology, when we have non-ideal geometries, what are the two other types of geometries that exist in coordination chemistry?
Pseudo-geometries: Geometries minorly deviating from idealized shapes. Deviations are often due to minor steric or electronic effects.
Distorted geometries: Geometries majorly deviating from idealized shapes. Deviations are often due to major electronic effects, e.g. Jahn-Teller.
Are d-electrons stereochemically active? Do they still have influence on shape? Are they the only factor determining shape?
Even if d-electrons are not stereochemically active, they still can influence shape, but they are still not the only factors for shape.
What does crystal field theory (CFT) state?
What does ligand field theory (LFT) state?
Theory stating that ligands, acting as point charges positioned at fixed spots around the “field,” interact with the metals differently because of where the metals’ orbitals point at.
The theory that combines concepts of crystal-field theory and molecular orbital theory, describing bonding in terms of interactions between d orbitals of metals and the frontier orbitals of ligands giving birth to molecular orbitals.
In an octahedral ligand field, what orbitals are the ones directly pointing at the point charges or the ligands?
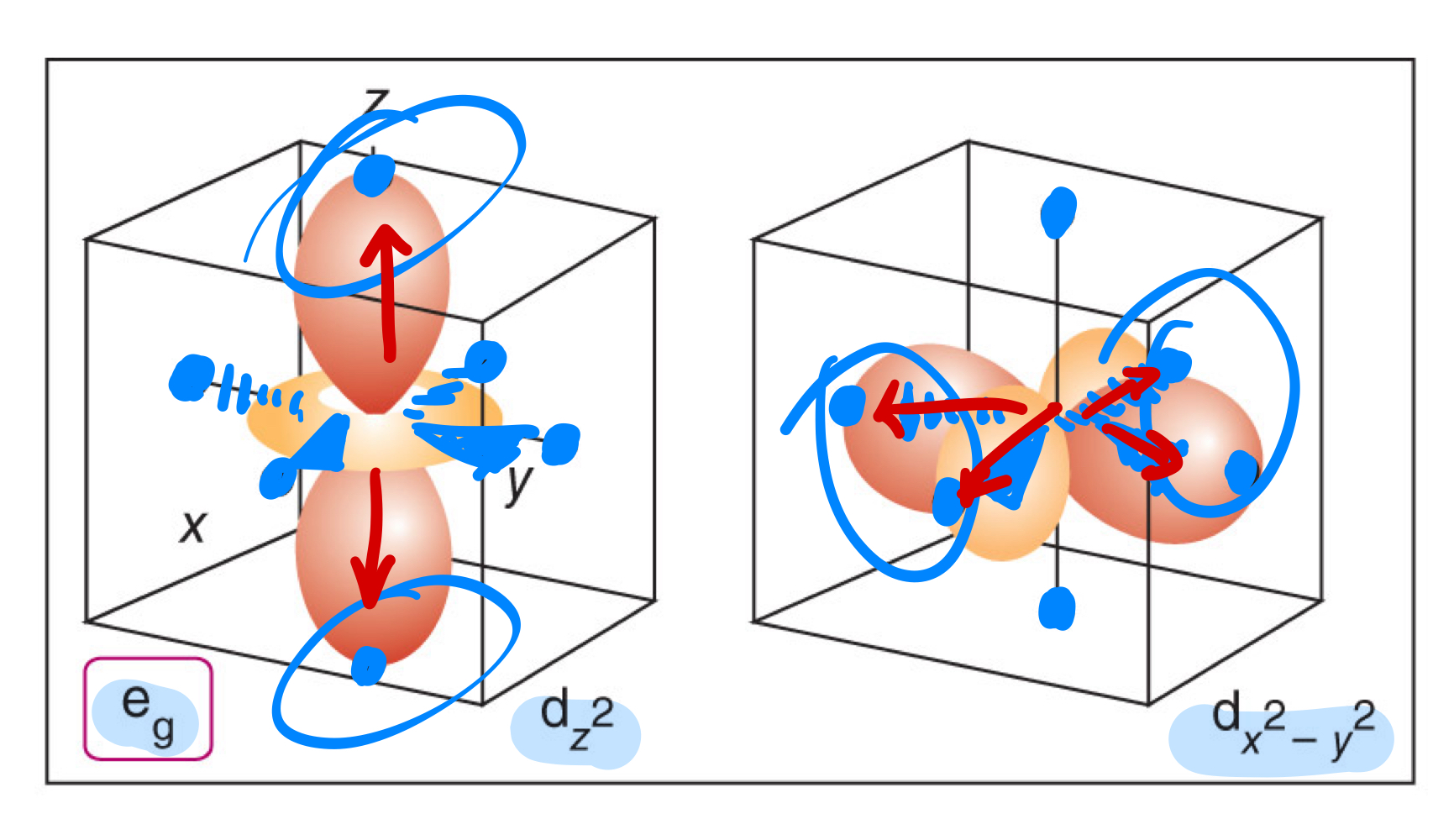
In an octahedral ligand field, what orbitals are NOT the ones directly pointing at the point charges or the ligands?
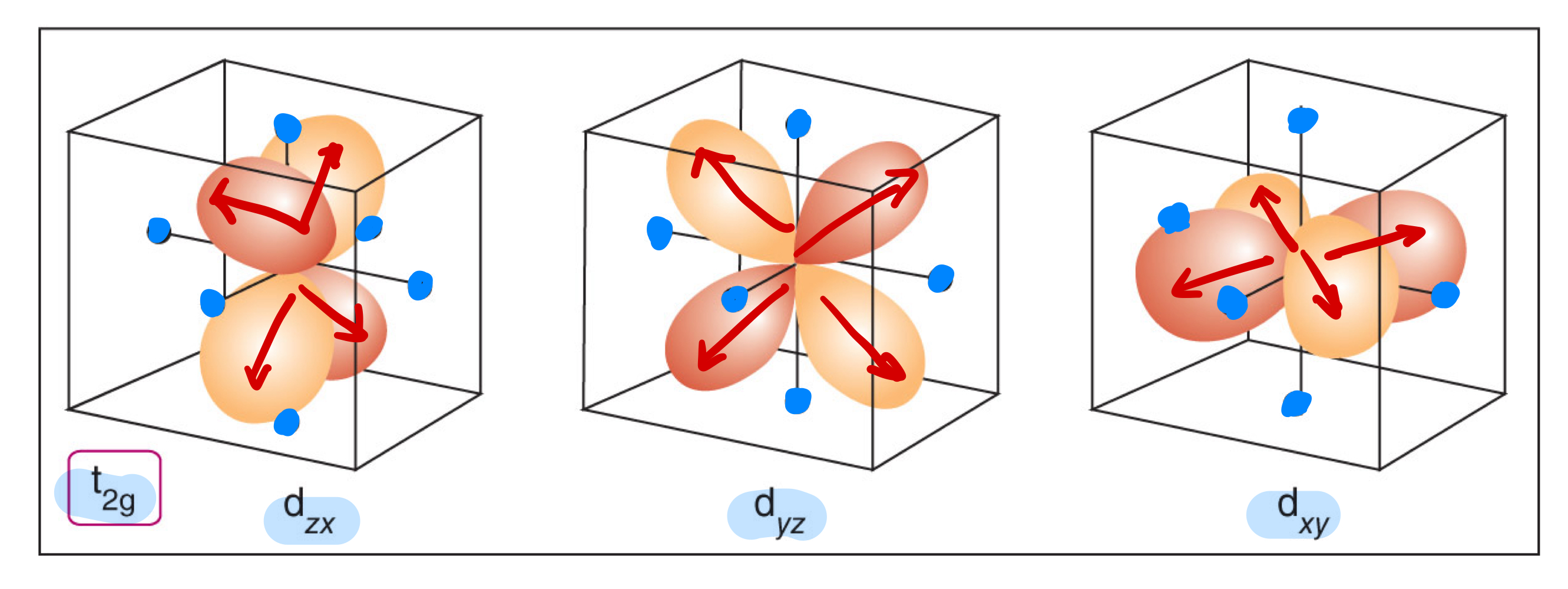
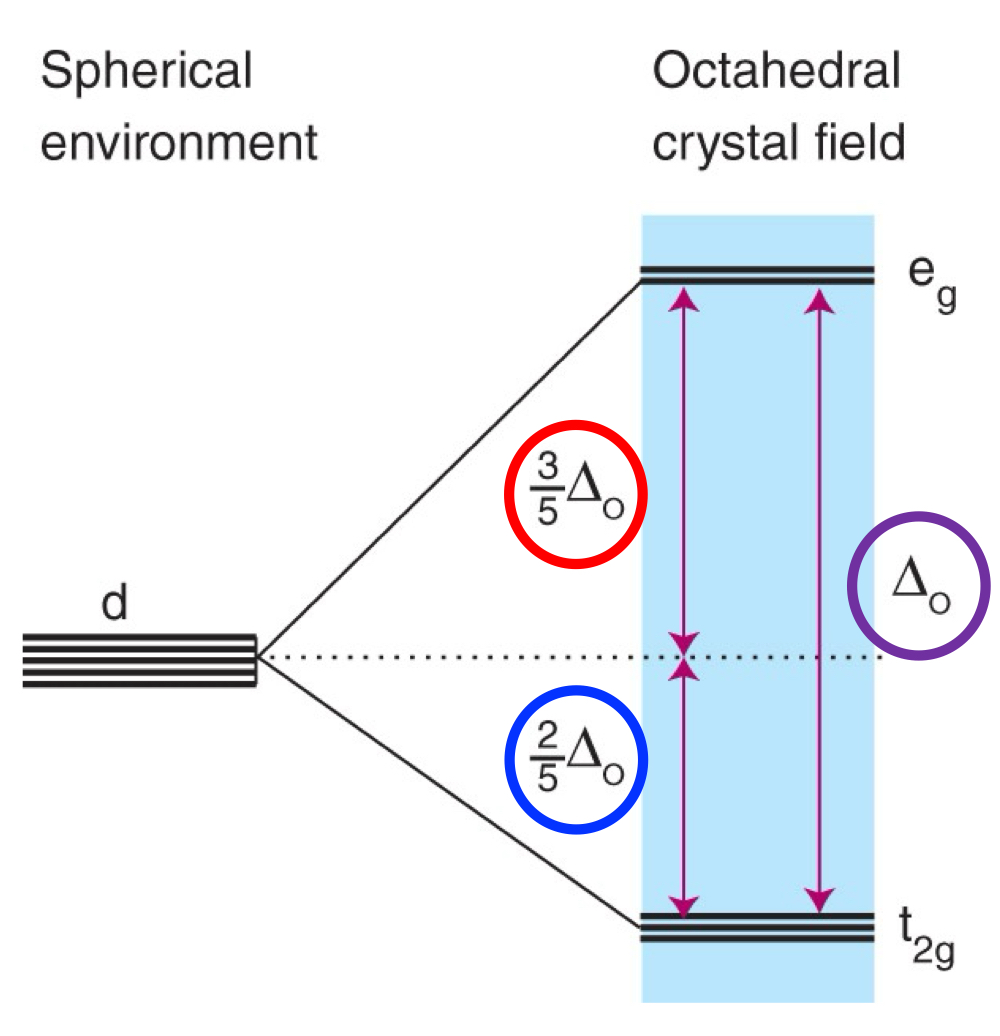
What is the ligand-field splitting parameter (∆o)?
What is the barycentre?
What would change ∆o with?
What is the spectrochemical series?
The energy required to move electrons between these two sets of orbitals.
The energy that all five d orbitals would have in a hypothetical spherically symmetric environment wherein the “negative charge” of the ligands are everywhere in the sphere.
∆o would change with the identity of the ligand and metal centre.
A series of ligands based on increasing ligand ability to cause a splitting of d-orbitals in transition metal complexes.
What does the spectrochemical series increase from and in? Hint: Two ways of analyzing this trend.
From small ∆o to big ∆o:
π-donors to π-neutral to π-acceptors.
Hard bases to give to soft bases to give.
Why do strong field ligands create a bigger ∆o?
Why do weak field ligands create a smaller ∆o?
Strong field ligands create a bigger split because soft bases and π-acceptors do a good job in donating electron density into the d orbitals, causing repulsion, thus requiring more energy to populate electrons in the metal orbitals.
Weak field ligands create a smaller split because hard bases don’t do a relatively good job in donating electron density and π-donors withdraw electron density from the d orbitals, both cases not creating that much repulsion, thus requiring less energy to populate electrons in the metal orbitals.
When you increase the oxidation state of a metal, what happens to the size of ∆o?
When you go down a group for the metal in the complex, what happens to the size of ∆o?
Increase of oxidation state → Removal of electron density → Shortening of ligand-metal interaction → Stronger interaction → Bigger ∆o
Down a group → bigger d orbitals → more ligand-metal overlap and interaction → bigger ∆o
What is the pairing energy?
What is ligand field stabilization energy, and what is its formula?
Energy required to pair an electron with another electron, keeping in mind about electron-electron repulsion.
Stabilization energy gained by the d-electrons in a particular electron configuration
Formula: LFSE = (# of electrons in t2g)(-2/5 ∆o) + (# of electrons in eg)(+3/5 ∆o) + (# of pairing events)P
What are the two options of placing another electron to the d orbitals?
Option 1: Low spin. Once you have singly filled all orbitals in the t2g set, you would pair the remaining electrons in these filled orbitals. Usually corresponds to strong field ligands, wherein |∆o| > |P|.
Option 2: High spin. Once you have singly filled all orbitals in the t2g set, you would move the remaining electrons to the empty eg orbitals. Usually corresponds to weak field ligands, wherein |P| > |∆o|.
Where will the electrons of a metal ion with strong field ligands go? The t2g or eg, and why?
Where will the electrons of a metal ion with weak field ligands go? The t2g or eg, and why?
The metal’s electrons will go to the t2g orbitals (orbitals lower in energy), because why would I want to spend energy to climb a mountain when I can settle in its comfy trough? Low spin.
The metal’s electrons will go to the eg orbitals (orbitals higher in energy), because why would I not want to spend energy on a not-so-tiring hike up the mountain to get a good view? High spin.
For 3d metal ions, do they tend to be low-spin or high-spin with strong-field ligands? How about with weak-field ligands?
For 4d and 5d metal ions, do they tend to be low-spin or high-spin with strong-field ligands? How about with weak-field ligands? What explains the possible difference between these metals and 3d metals?
3d metal ions tend to be low-spin with strong-field ligands and high-spin with weaker-field ligands.
4d and 5d metals tend to be low-spin regardless if it’s strong- or weak-field ligand. It has bigger d orbitals, and so ∆o increases, which makes the case for low-spin. Additionally, it has small pairing energy due to more space in these orbitals causing less repulsion between to-be-paired electrons, causing the relative comfort of pairing electrons.
What’s the difference between dimagnetic and paramagnetic materials?
Diamagnetic materials have only paired electrons, paired spins, and are slightly repelled by magnetic fields.
Paramagnetic materials have some unpaired spins and are attracted by magnetic fields.
What does the Jahn-Teller theorem state?
Any non-linear molecule with will undergo a geometrical distortion that removes the degeneracy of unequally filled orbitals to lower the total energy of that molecule.
In a tetrahedral crystal field, what are the d orbitals that does point toward the ligands and destabilize the complex?
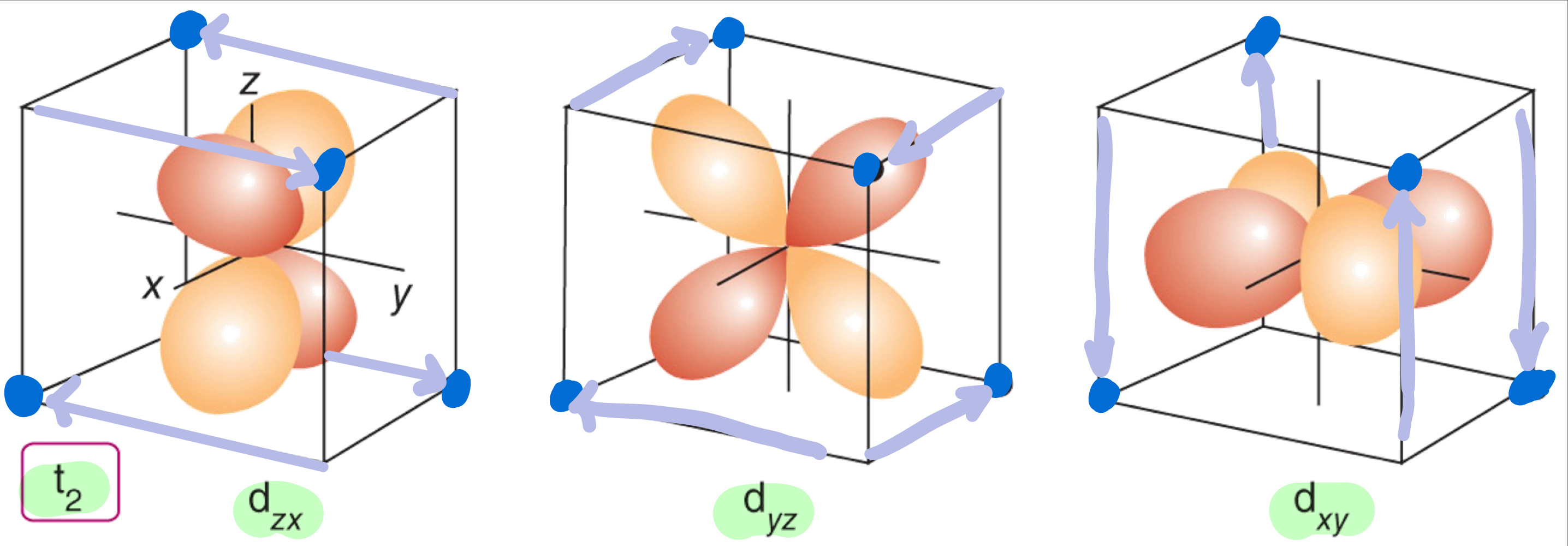
In a tetrahedral crystal field, what are the d orbitals that do not point toward the ligands and destabilize the complex?
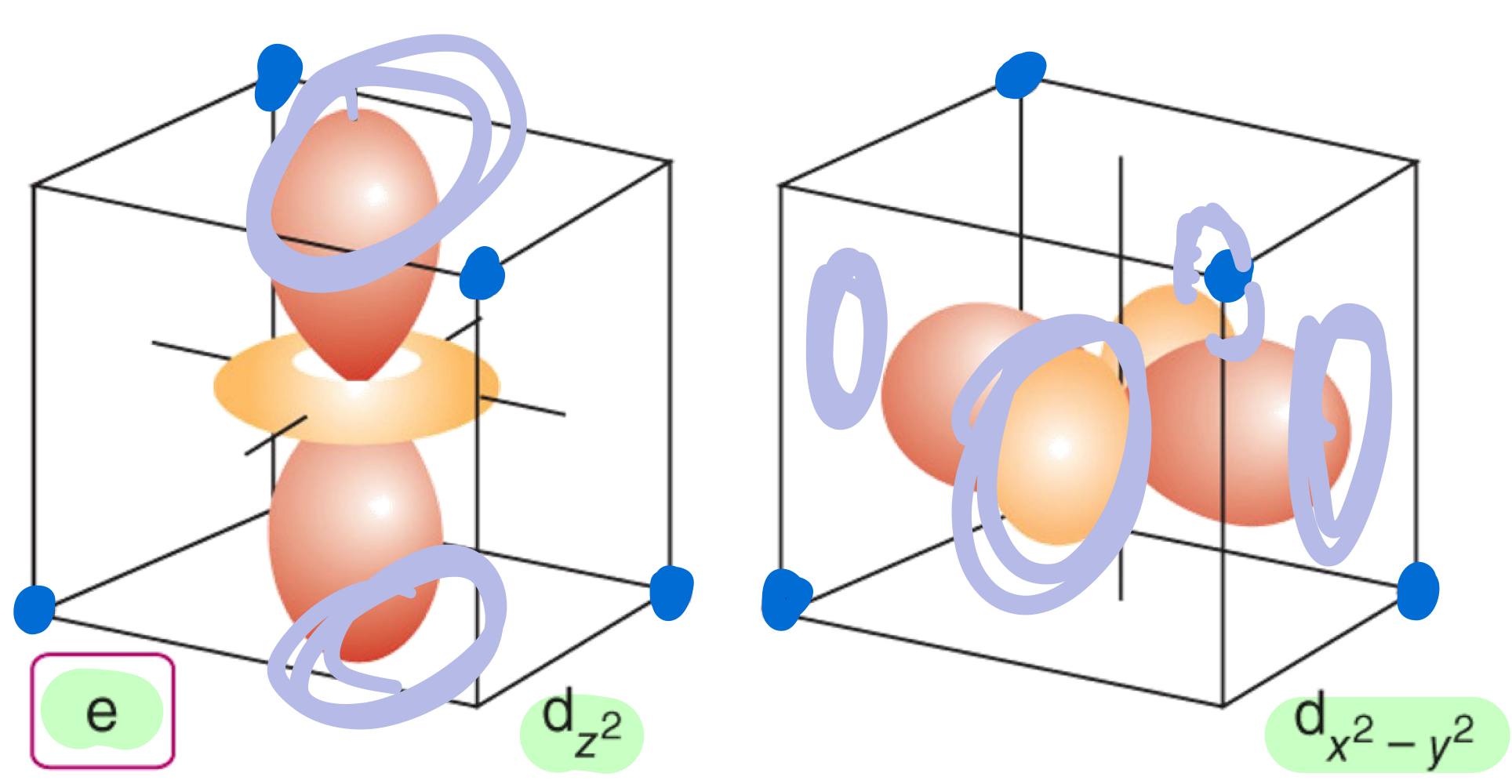
Why is LFSE smaller for tetrahedral ligand field than octahedral ligand field?
The d orbitals interact less strongly with the ligands in a tetrahedral arrangement
There are only four negatively-charged regions rather than six.
For a particular complex between a metal and a ligand, what’s the ratio of ∆t and ∆o?
1 ∆T = 4/9 ∆o, wherein |P| is generally larger than |∆T| because of this.
Why do most tetrahedral complexes have high spin?
Td complexes are more likely to be high spin because the “higher energy orbitals” t2 set are less “high in energy” than the anti-bonding eg set in Oh. Because these orbitals are more accessible, it’s less energetically costly to put electrons in them than to pair them in the lower energy manifold. Thus, the “high spin” configuration can usually be the ground-state (i.e., lower in energy) electronic configuration.
For high-spin octahedral or tetrahedral complexes, there is no LFSE with empty, half-full, or full d shells. Why?
Because they all calculate to 0. There is not stabilization.
Why would d8 metals with strong ligand fields favour square planar over tetrahedral?
Because of the stabilization it has by lowering energies of atomic orbitals, not ligand-group orbitals anymore.
What are the three criteria for a 4-coordinate complex to be square planar? Use the following: d system, ligand field strengths, and crystal field splittings.
d8 metals with strong field ligands and/or bigger metals with strong crystal field splittings.
What is it about being a d8 system (with strong field ligands and/or bigger metals with strong crystal field splittings) that increase the chance of being square planar instead of Td?
The idea here is that strong ligand fields provide an electronic driving force for adopting SP configurations; the big gap made between the dx2-y2 and the rest of the d-manifold makes it more stable but for electronic reasons.
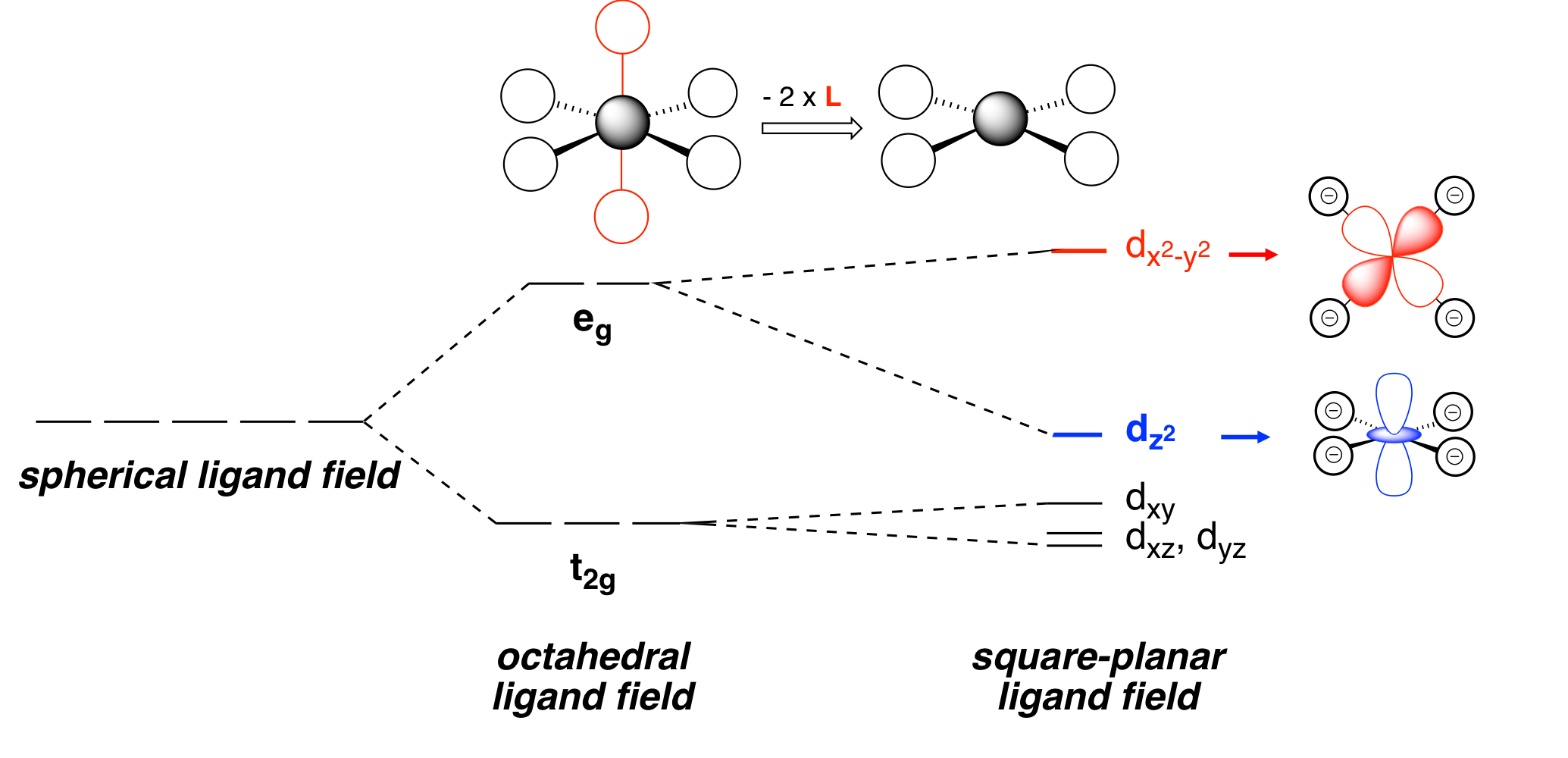
Explain the new orbital energy changes/splittings from an Oh ligand field to SP` ligand field.
dz2 decreases in energy because now that the axial ligands are removed = no repulsion = less energy.
dxz and dyz because these are orbitals that are on the z-axis, which is the same axis where the axial ligands were. Now that you removed them, similar reasoning with dz2 = no repulsion = less energy. Less drastic than dz2, due to where the electrons are positioned.
dxy and dx2-y2 goes up in energy because when you remove the axial ligands, the equatorial ligands will move closer to the metal in such a fashion that it will end up interacting with the dxy lobes, causing repulsion = more energy. The equatorial ligands and axial ligands used to repel each other, rejecting the equatorial ligands to get closer to the region where the dxy lobes once were. Now that the axial ligands are gone, they can get closer to that region and now repulsion occurs.
If all ligands were the same, what is the ordering of z2, xy, xz, and yz only dependent on? What if the ligands were not all the same?
If all the ligands are the same, The ordering of z2, xy, xz, yz only dependent on the pi-accepting ability of the ligand.
If the ligands are not the same, the ordering can be dependent on other factors.
What explains the preference for d8 SP with bigger metal centres and stronger field ligands?
What happens if we had a small metal and weak field ligand?
Due to the interaction of ligand and metal leading to increased ∆. If that’s so, small metal and weak field ligand = smaller ∆ which would cause Td.
What dictates whether a 4-coordinate complex will adopt a SP or Td geometry?
Which geometry results in a lower overall energy.
Why do we have to pay attention to the d-electrons of the metal in the grand scheme of ligand field theory?
The d electrons are where a complex’s properties and reactivity come from - specifically because they are tied to the frontier orbitals (HOMO and LUMO) as the t2g and eg* sets.