medchem exam 2 ADMET 1
1/61
Earn XP
Description and Tags
working on march 26th class, class 9, all done
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
62 Terms
drug pharmacodynamics descr
interaction w/ receptors, enzymes, lipids, DNA; the study of the biochemical, physiological, and molecular effects of drugs on the body; what a drug does to the body
pharmacokinetics descr
describes what the body does to a drug, analyzing its journey through ADME; how to get to targets, stability
toxicology descr very basic
safety
ADMET long
absorption, distribution, metabolism, excretion, toxicology
what is the Cmax?
highest concentration of a drug in the blood/CSF/target organ after a dose is given
this that affect how a drug is administered (3)
patient convenience, local action, drug properties
ways that drugs are administered (just list them) (2, 4)
oral: inhaler, pill
through skin: intravenous (into vein), intramuscular (into muscle), subcutaneous (layer of tissue beneath dermis/epidermis), transdermal (like a skin patch)
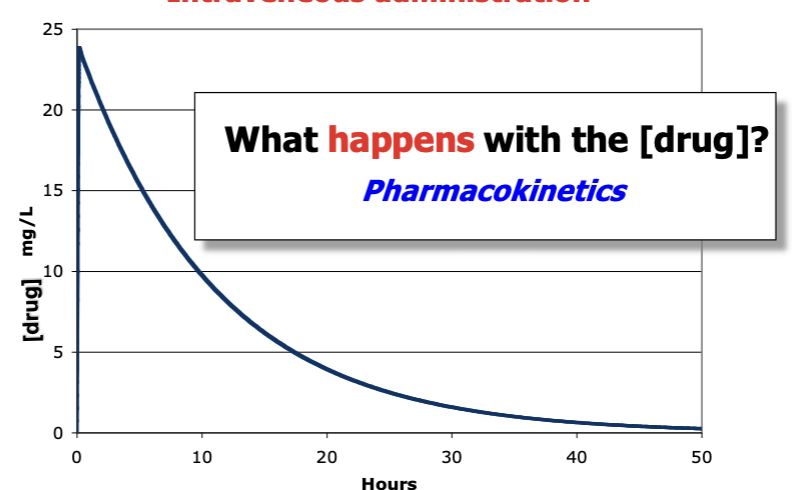
what type of administration is this? + descr
intravenous - concentration gets the full dose first and then decreases over time
oral administration descr
slower uptake- first a slower increase (lag time) then a slower decrease
therapeutic window descr
space between minimal effective [] and the place of adverse high effects
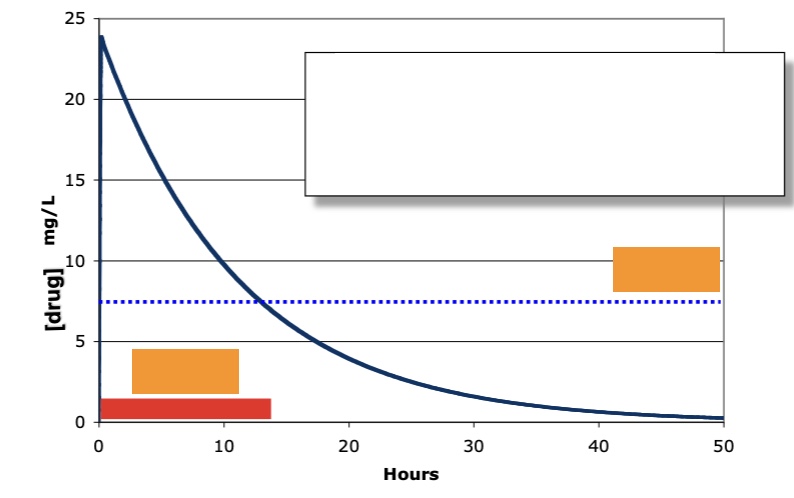
IV administration: label the orange (red (what is on the slide, not added by me) and blue)
red: biologically active (window of time when it is)
blue: minimal effective [] (MEC)
uptake, breakdown and elimination for drugs (what parts of ADMET are they)
uptake: absorption
breakdown: metabolism
elimination: excretion
half life def
time it takes to go to 50% of [drug] from max
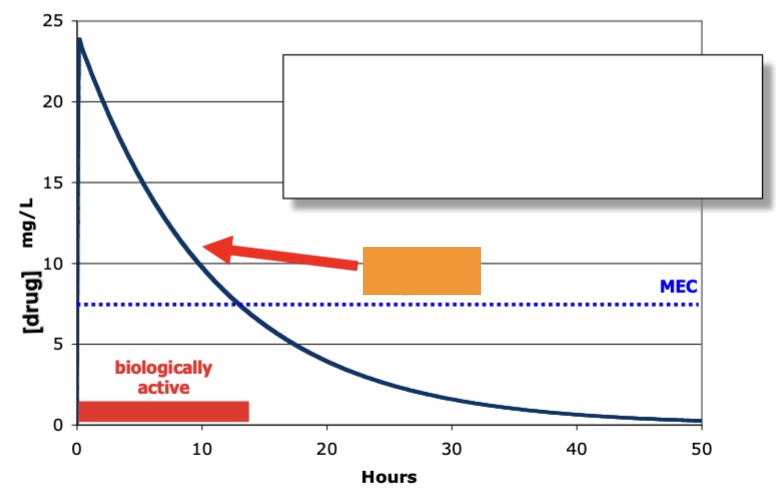
label orange
half life (t1/2)
half life egs for aspirine and ibuprofen
asprine: 0.38 hrs
ibuprogen: 2 hrs
area under curve def
total amount of the drug in circulation
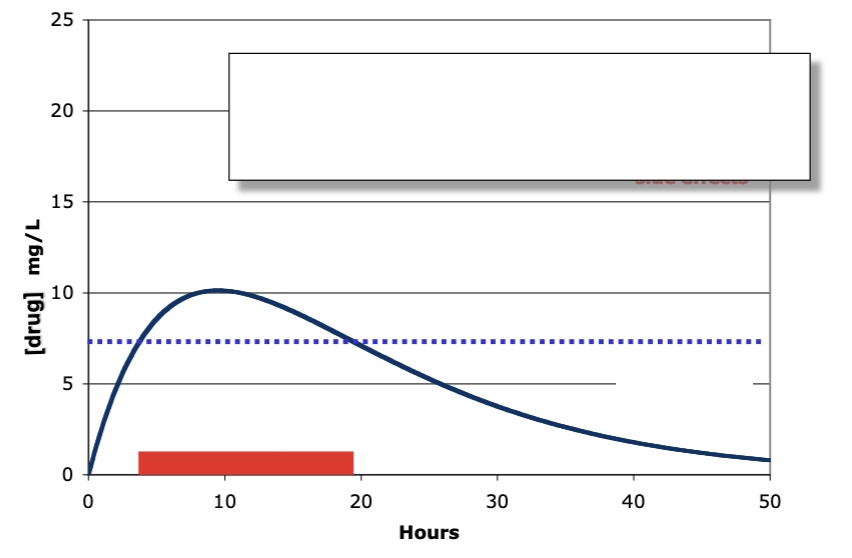
what type of administration, what is the red bar, what is blue line
oral administration
red: pharmacological activity
blue: MEC
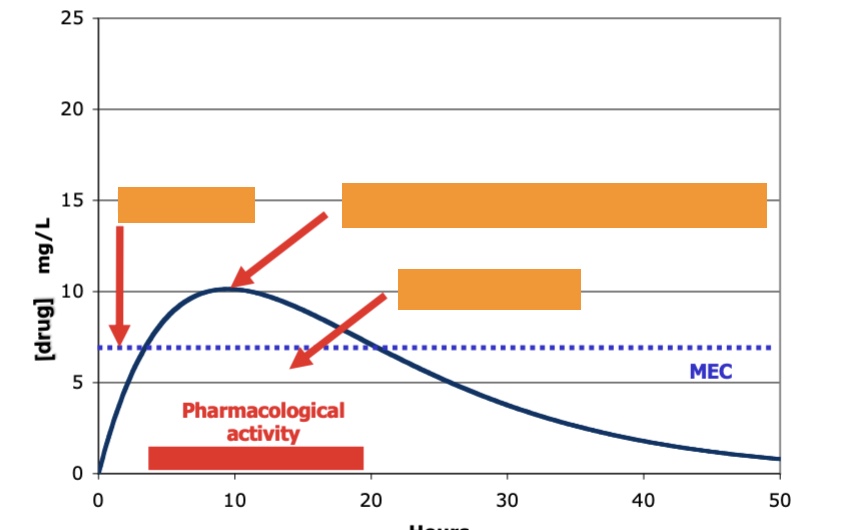
label left to right, oral administration
lag time, Cmax (relates to adverse drug reactions), Area under curve (from time 0 extrapolated to infinity)
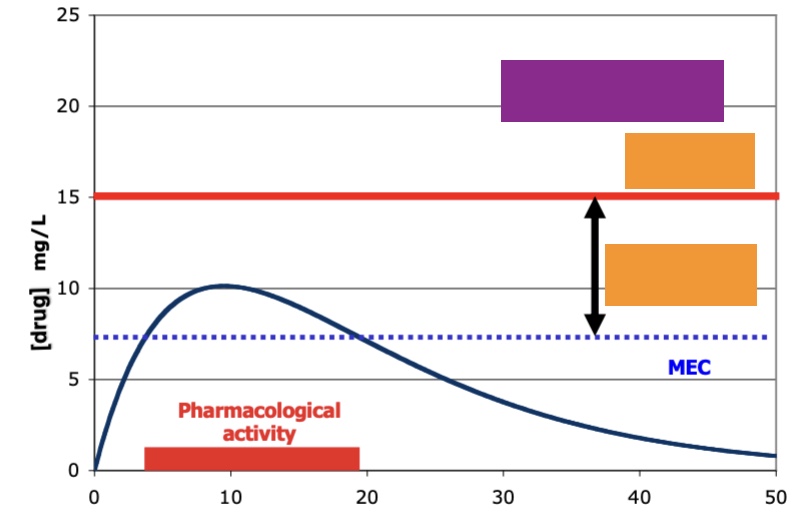
label top to bottom (purple is the field that covers the ff orange)
purp: toxicology
orange: adverse side effects, therapeutic window
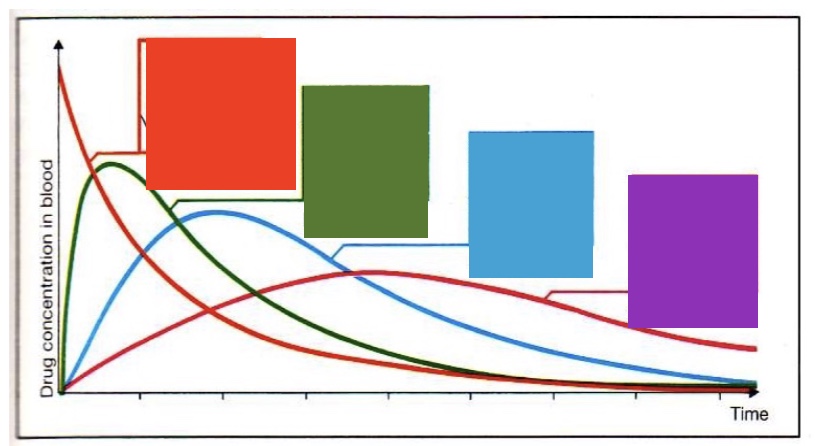
label left to right - what drug [] curves do diff routes of administration provide
intravenous, intramuscular, subcutaneous, oral
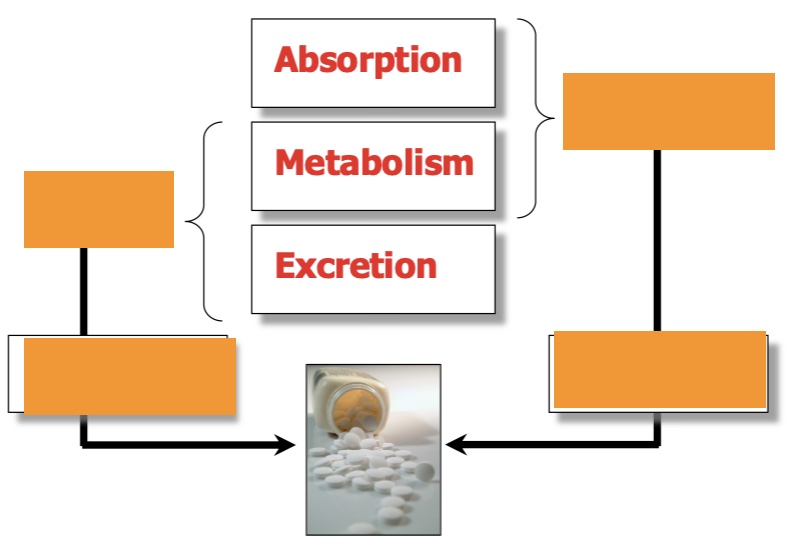
label left top then bottom, then right
drug half life, how (often)?
oral bioavailability, how much?
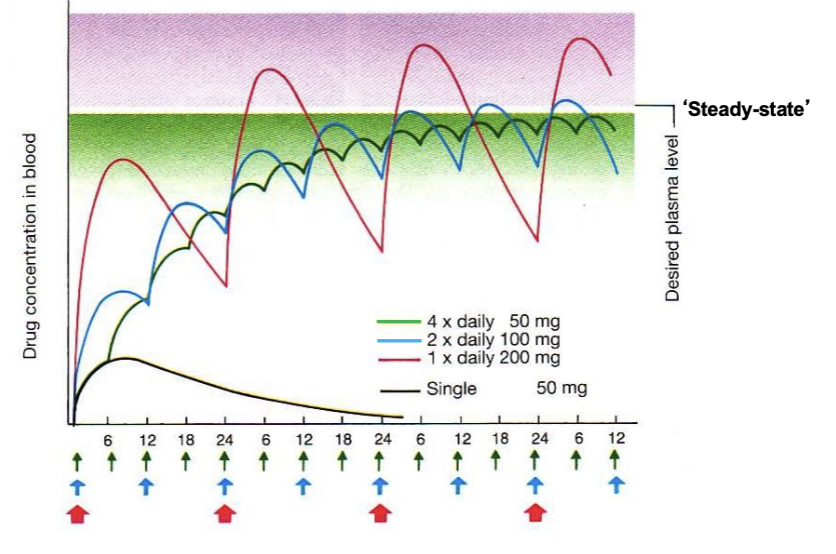
what does this image show?
in real life - often repeated oral dosing to get desired plasma [] (each color is a diff drug)
reasons for clinical failures of drugs in order of importance (3) (in 1988)
pharmacokinetics (most important!! 39% !), lack of efficacy, animal toxicity
what does the uptake of a drug depend on
rate of absorption - differs by indiv, what they ate, what transporters in gut, etc.
how how the percentage that ADMET affects successfulness of clinical trial changed 1988 to 2004
decreased significantly (from 39% to 8%)
first hurdle for oral dose
absorption, goes through the liver first
what is very important from pill to drug (4 points)
solubility in water!! determined by drugs ability to interact w/ water molecules
uncharged - H-bond donors (HBD), H bond acceptors (HBA)
charged - dipoles (H2O)
variability between chemicals is large
solubility of a drug what it means
>65 microgram/mL solubility not limiting absorption
<10 microgram/mL no absorption
hence candidate drug is often required to have solubility above 10 to facilitate preclinical testing
what is important for charged molecules to be soluble in water
ionization
what is important for charged molecules to be membrane permeable
having neutral species (non ionized)
absorption and molecular size (3)
large molecules need enough functional groups to be soluble, functional groups from hydrogen bonds w/ water molecules → solvation shell, to penetrate liquid membrane solvation shell needs to be disrupted
what does the MV have to be for it to be incompletely absorbed
MV > 500
Lipinski’s Rule of 5
one of the big dogmas for drug-like molecules
MW < 500, Log P < 5 (does compound prefer water of hydrophobic context, partition constant), H-bond donors < 5 (sum of OH and NHs), H bond acceptors < 10 (sum of N and O atoms w/ free pairs)
Log P descr
experimental measure of hydrophobicity
internal transport mechanisms after oral intake
from intestinal lumen, through gut epithelial cells with active transport; transporters really important!! diff transporters on each side cause polarized cells
4 diff types of transporters
amino acid transporter, oligopeptide, phosphate transporter, glucose transporter - we know specific examples for each
P-glycoprotein descr
“bad guy” - blocking it helps w/ absorption, uptake depends on how many you have; BUT involved in keeping foreign molecules out of the brain (BBB), also often upregulated in cancers
next hurdle after absorption
first pass effect - an orally administered drug is metabolized by the liver, intestines, or lungs, significantly reducing its active concentration before it reaches systemic circulation; impacts bioavailability
balance of drug uptake two ways
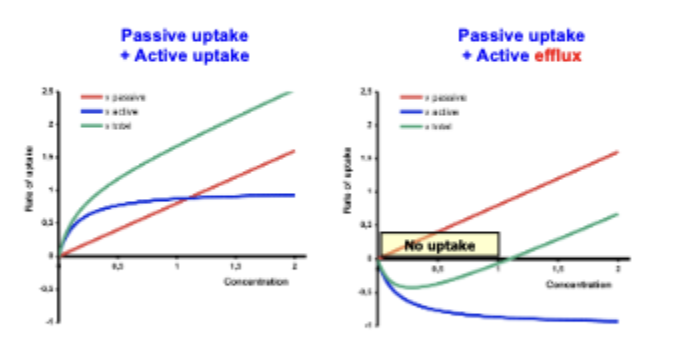
what else can happen w/ active transport and drug efflux
drug-drug interactions
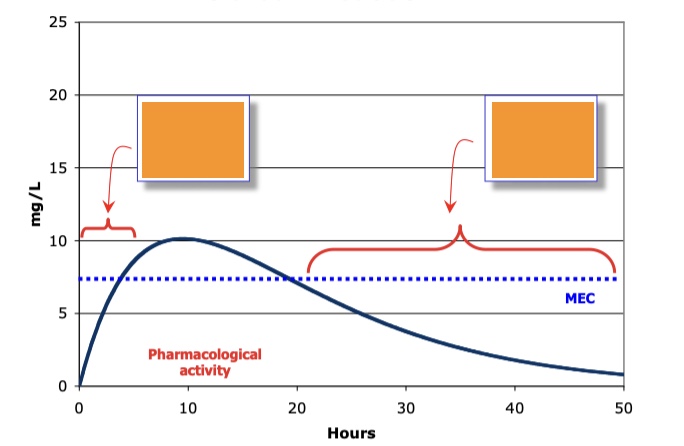
label
absorption phase (first pass metabolism); elimination phase - metabolism excretion
first pass effect by the liver
metabolism by enzymes, like the cytochrome P450 family
paracetamol metabolism (2)
phase 1: CYP2E1, phase I reaction enzymes, after uptake by cell; phase 2: conjugation, using phase II reaction enzymes
what is drug metabolism
done by specialized enzymes, in order to make the drugs more water-soluble and ready to excrete via the kidneys, liver is the main site of metabolism for most drugs (also a bit in the GI, lungs)
metabolic stability def
determines the half life of a drug, important! Involves the inactivation of an active drug, activation of inactive drug (prodrug), increased excretion of more polar compounds
microbiome role
matters with ADMET quite a bit!!
metabolism/biotransformation descr (3 steps + what in between, egs)
lipophillic drug -(phase I- reactions, eg oxidation, reduction, hydrolysis; by CYP)→ functionalized drug (drug-XH, metabolite (has a OH group)) -(phase II rxns, eg glucoronidation, sulphation; conjugation (?)) → conjugated drug (makes it so that it is very water soluble and can be excreted by the kidneys)
eg of a phase I enzyme
Cytochrome P450
eg of phase II enzyme
UDP
CYP expl
haem enzyme - has a haem group which has an iron molecule in between; the iron molecule can activate oxygen which can oxidize what is inside it if it fits
what is drug binding to CYPs like?
drugs binding to their targets - about molecular interactions and making the right fit
what can N-containing groups act as for CYP?
CYP inhibitors
major isoforms for drug metabolism
2C9, 2D6, 3A4 - some people lack them and then some drugs dont work on them
variation in metabolism
diff people have diff CYP, depends on diet, genes, etc. can change over time too - why people react differently to different drugs
next part of drugs life
excretion - kidney goes into urine, liver and GI goes to faeces and bile
kidney basic numbers
180L/day filtered, excreted 2L/day
importance of plasma protein binding
blasma is 55-60% of blood volume, contains proteins that can bind drugs, eg albumin can bind weak acidic drugs, alpha1-Acid glycoprotein can bind basic (cationic drugs)
drug-drug interactions eg
warfarine - anti-coagulant w/ v low therapeutic window, v strong albumin binding
sulphonamide abx - very strong albumin binding, competes w/ warfarine leading to bleeding by high levels of free warfarine
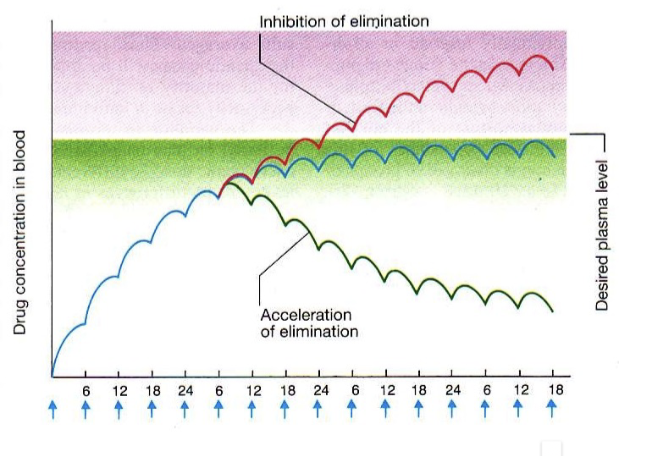
what does this show
how diff drug drug interactions can effect drug concentration in blood
grapefruit juice and drugs
juice contains inhibitor for CYP3A4
side effects w/ drugs (3)
often via interaction with other proteins, depends on used drug [], could also be due to metabolites
phase I and phase II descr
drug can be metabolized directly by phase I OR metabolized by phase I then II