All of concepts exams so far combined
1/155
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
156 Terms
Radiation that hits earth’s surface
UV, visible, IR (heat)
Greenhouse effect
earth emits longer IR wavelengths
Which are absorbed by greenhouse gases → warming atmosphere (essential for habitability)
GG’s remit same E IR in random places (space, earth again → global warming)
Greenhouse gases
gases that absorb IR when photon w/ same E strikes molecule → excited state, Re-emits photn of same E in random directions
To be a greenhouse gas
molecule must absorb at least 1 wavelength of IR released by earth
Have at least 1 polar bond
Bond types in order of inc. En
nonpolar < polar covalent (permanent dipoles) < Ionic
What determines molecule polarity
bond polarity + molecular geometry (determines if overall molecule is polar/non-polar)
Covalent bonds
not rigid, vibrate
Fluctuating molecular dipoles
Permanent or temporary
IR active
Creates charge changes
3 types of fluctuating molecular dipoles
Direction of dipole changes
Magnitude of dipole changes
Temporary molecular dipole appears and disappears
Dipoles
Polar → permanent
Non-polar → temporary, will only have temporary dipoles when vibrating if contains polar bonds
*non-polar bonds → typically IR inactive
IR active
Vibration → charge fluctuation, E of wavelength absorbed depends on vibration type → bending vs. stretching
Stronger the bond (“spring”), the more E needed to vibrate it
All molecules w/ polar bond have at least 1 vibration which changes molecular dipole
Relationship between transmittance and absorbance
lower transmittance = greater absorbance
CO2 capture
Capture at source
DAC → direct air capture
*as atmospheric CO2 inc, [ ] of dissolved CO2 inc. in oceans → acidification
![<ul><li><p><span style="background-color: transparent;">Capture at source </span></p></li><li><p><span style="background-color: transparent;">DAC → direct air capture </span></p></li></ul><p><span style="background-color: transparent;">*as atmospheric CO2 inc, [ ] of dissolved CO2 inc. in oceans → acidification</span></p>](https://knowt-user-attachments.s3.amazonaws.com/4fae3f6f-dc55-4cda-8140-dc598dd2456f.png)
C neutral
no change in amount of CO2 in atmosphere, use fuels w/ less C like MeOH
Can be achieved w/ carbon capture
C (-)
decreases amount of CO2 in atmosphere
Can be achieved w/ carbon capture
C(+)
inc. CO2 in atmosphere → world currently
Solubility of gases in liquids
Pgas impacts solubility of a gas in a liquid, Cgas= gas solubility - max [ ] of dissolved gas
Henry’s law
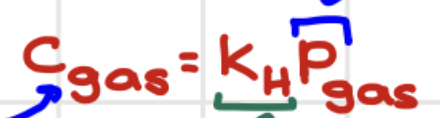
Henry’s law constant (KH)
is different for each particular gas
Polar gases have > KH
CO2 has greater KH than other nonpolar gases ( it reacts w/ water to make carbonic acid which in turn produces more aqueous CO2)
Partial pressure (Pgas)
the pressure exerted by an individual gas in a mixture
Is equal to the pressure that the gas would exert if it were all by itself
Cgas is proportional to Pgas
When partial pressure of gas above soln inc., solubility of gas also inc.
Dalton’s law of partial pressures
the P of a mixture of gases is the sum of the partial pressures of component gases
Mole fraction (x)
a unit of [ ] defined as # of moles of a component in a mixture divided by the total # of moles of all components
![<p><span style="background-color: transparent;">a unit of [ ] defined as # of moles of a component in a mixture divided by the total # of moles of all components</span></p>](https://knowt-user-attachments.s3.amazonaws.com/2e57dfd9-d7e2-4c27-a76a-4c503e2f34d5.png)
Solubility impacts of gas in a liquid
Temp → inc. T, dec. Cg, inc. KH
Polarity → like dissolves like
Le Chatliers principle
when a system at equilibrium is disturbed (or stressed), it responds by re-establishing equil. to reduce applied stress
Adding aqueous or gas phase reactant/pdt to a system
system proceeds in direction that consumes part of the added species
Removing aqueous or gas phase reactant or pdt to a system
shifts system in direction that restores part of the removed species
How oceans capture CO2
ocean acidification
Bronsted-lowry model
involves H+ transfer, good for showing what happens in rxn
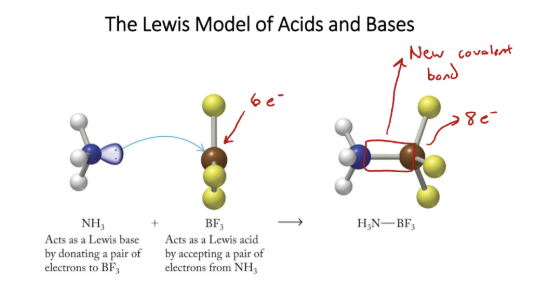
Lewis model
shows transfer of e-, good for showing what happens
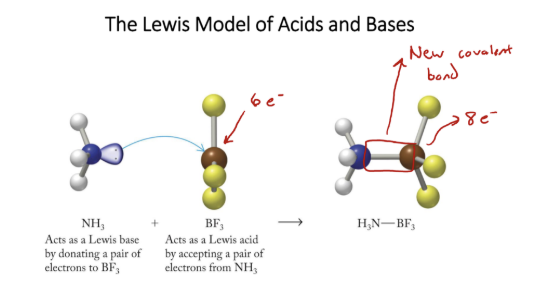
Lewis acid
accepts lone pair, cations, neutral molecules with partial + charges
Lewis base
donates lone pair, most often anions or neutral molecules with LP’s
How MOF’s capture CO2
Physioabsorption → absorption by IMFs
Chemisorption → formation of chemical bonds
* indicator → HIn = conj. A, :In- = conj :B
Infared spectrum for CO2
Asymmetric stretching = 4.25 µm → more E, smaller wavelength
Bending = 18 µm → less E, longer wavelength
3 vibrational modes for CO2
Symmetric stretch → IR INACTIVE (no fluctuation in net dipole)
Asymmetric stretch → IR ACTIVE
Bending → IR ACTIVE
Stretch = change in bond length
Bending = change in bond angle
3 vibrational modes for H2O
3 peaks → therefore ALL IR ACTIVE
Asymmetric stretch
Symmetric stretch
Bend
Why is methane a greenhouse gas
although seen as nonpolar in most situations, it is IR active. Also, any time bonded atoms are different, consider molecule to be non-polar and IR active, its tetrahedral structure allows for C-H vibrate at frequencies that match certain wavelengths of IR radiation
Wave number (ν̃)
another unit on IR graphs, low ν̃ = low E and therefore long wavelength and vice versa
CO2 → carbonic acid rxn
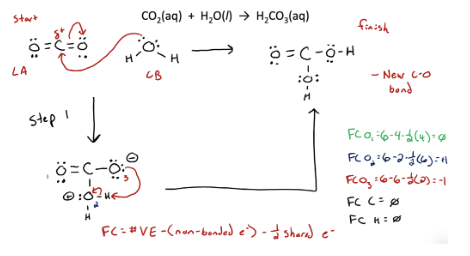
CO2 → K2CO3 rxn
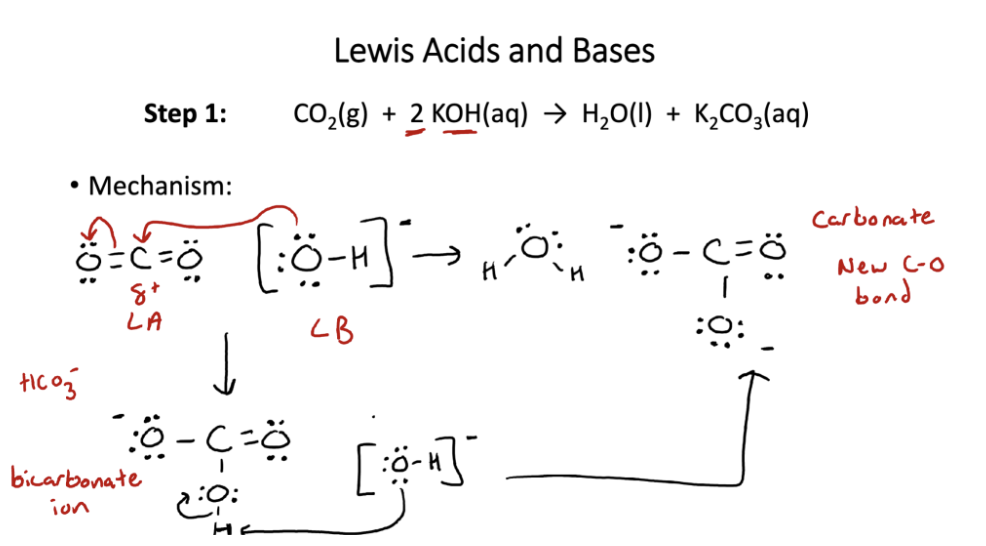
Why do no other abundant atmospheric gases bind to -OH

Sequestration
storage of CO2
Terrestrial → trees
Geological → underground
T and P At surface level
Pressure = 1 atm
Temperature = 298K
Ideal gas law
PV=nRT
R=0.08206 (L*atm)/mol*K
Ideal Gases
Ideal gas law is accurate at reg, T’s and P’s
Calc. value agrees with experimental/actual value
Becomes inaccurate at ↑P and ↓T
Assumptions based on Kinetic Molecular Theory (KMT)
Assumptions:
Conditions are not typical or accurate underground w/ higher P
Gas particles = point masses = negligible volume
G parts move constantly and randomly throughout space they occupy, elastic collisions, collisions w/ container walls is origin of gas pressure (each collision produces a tiny force)
G’s have negligible IMFs (particles are far apart) (we assume that g particles don’t interact)
KEavg of gas molecules ⍺ T (in K), ∴ ↑T, ↑KEavg, m = const, urms must ↑
T ⍺ V, rises underground
Kinetic Energy
KE = ½ mu^2
KE avg is used to describe gas particles (not all particles move at same speed)
Rms = speed of particle w/ KE=to KEavg of all particles in a gas
KEavg = ½ mu^2rms
* urms ↑ w/ ↓MM
At same T all gases have same KE avg, ↓m, urms must ↑
One mol of ideal gas if P doubles
volume decreases by 50% ↑P, ↓V
At high P
1) Gas particles are closer together ∴ IMFs are no longer negligible
These actions:
reduce the frequency of particle collisions w/ container
reduce force of particle collisions w/ container
∴ less particles moving more slowly
PV/RT term is lower at same T for molecules with more IMFs
2) V of gas particles themselves is sig. Fraction of total V occupied by the gas
Gas sample isn’t all “empty space”, particles are not compressible and don't shrink w/ increasing P
∴ actual volume is greater than expected
Rigid walls at High P
Larger volume = particles don’t interact much, behave like an ideal gas
Smaller volume = particles interact more, IMF’s cause particles to stick, act. P is lower than expected
Flexible walls at high P
at higher external pressure, particles interact more
Actual volume is higher than expected
Low T
G’s deviate from ideal behavior
↓T = ↓KEavg bc KE ⍺ T
Causes particles to “stick” together
reduce the frequency of particle collisions w/ container
reduce force of particle collisions w/ container
Rigid walls at lower temp
actual P is lower than expected
Flexible walls at lower T
the actual volume is lower than expected
Van der Waals Eqn./Real gas law
Constants:
a = increases as IMFs increase
b = increases as size increases
a(n/vactual)² corrects for the fact that Pactual is lower than that predicted by the ideal gas law due to IMFS
b corrects for the fact that Vactual is greater than predicted by ideal gas law due to the size of the particles
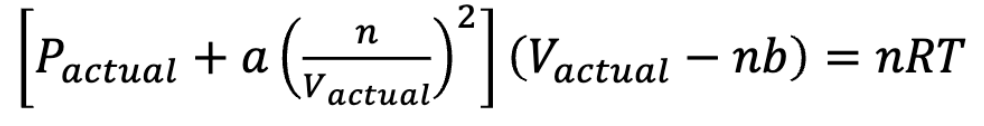
forming a real gas order
ideal gas → real gas → liquid
Rule for P underground
increases by about 100 atm/km
Rule for T underground
changes very little for first few hundred meters until about 300m, then rises by 25℃/km
Pumping CO2 underground
↑P, ↑T
Volumes of gas samples decrease because ↑P, ↓V, although T is ⍺ to V, higher P dominates
At low enough depths CO2 becomes supercritical fluid
Phase diagrams
X-axis = T, y=P
Depend on 3 factors:
IMF strength
T
P = ratio of forces to SA, Pext impacts how close particles are, impacts phase of sub.
Heating s→l: KE is high enough to overcome some IMFs so solid melts
If KE is high enough it completely overcomes all IMFs and liquids can vaporize or solids sublime
blue = s → l
Red = l → g
G = s → g
Pressure
At higher P
Particles are closer together
Substance is more dense
At lower P
Particles are farther apart
Substance is less dense
For all sub.’s g phase has lowest density and is favored at lower pressure (l and s favored at higher P)
For all sub.’s boiling and sublimation T’s are greater at higher P (as water boils, more moles of gas above liquid, ↑n, ↑P above liquid)
Triple pt
all phases coexist
Supercrit fluid
has properties of both a liquid and a gas
Like a liquid: non-compressible, can dissolve stuff (CO2 = nonpolar solvent)
Like gas: ultralow viscosity, can diffuse though tiny cracks in some rocks
Super critical water has 10x more energy than hot water or steam
CO2 → Rock
M2+ w/ SiO4 4- (silicate)
Fe2+, Ca2+, Mg2+
M2+ reacts with CO3 2- (carbonate) to make MCO3
The MCO3 precipitate will form, storing the CO2 only if the [ ] of M2+ ions and CO32- are sufficiently high enough
How CO2 forms into rock
CO2 is acidic and dissolves underground metals releasing M2+
CO32- ions reacts with M2+ ions to make solid MCO3
H2CO3
polyprotic (diprotic) acid, donates 2 H+ ions, so has 2 Ka values, [H3O+] produced in 2nd step of rxn is negligible
Ka - ionization constant
The equil. Constant for the process of an acid ionizing in water
The higher the Ka value, better H+ donor, higher [H3O+], stronger the weak acid
pKa = -log Ka
↑Ka, ↓pKa
For polyprotic acids, why does the acid get progressively weaker?:
As the acid becomes more negative. More difficult to donate H+. HCO3- is ∴ a weaker acid than H2CO3 and has a lower Ka
*exclude liquids and solids when calculating
![<ul><li><p>The equil. Constant for the process of an acid ionizing in water</p></li><li><p>The higher the Ka value, better H+ donor, higher [H3O+], stronger the weak acid </p></li><li><p>pKa = -log Ka</p></li><li><p>↑Ka, ↓pKa</p></li></ul><p>For polyprotic acids, why does the acid get progressively weaker?: </p><p>As the acid becomes more negative. More difficult to donate H+. HCO3- is ∴ a weaker acid than H2CO3 and has a lower Ka</p><p><strong>*exclude liquids and solids when calculating</strong></p>](https://knowt-user-attachments.s3.amazonaws.com/9634adbe-b24d-4b02-a32c-f061a01d7110.png)
Kb - base ionization constant
The equil. Constant for the process of a base ionizing in water
↑Kb, stronger the base, higher [OH-]
pKb = -log Kb
↑Kb, ↓pKb
The acid with the lowest Ka will have the strongest conj. B
*exclude liquids and solids when calculating
![<ul><li><p>The equil. Constant for the process of a base ionizing in water </p></li><li><p></p></li><li><p>↑Kb, stronger the base, higher [OH-] </p></li><li><p>pKb = -log Kb </p></li><li><p>↑Kb, ↓pKb</p></li><li><p>The acid with the lowest Ka will have the strongest conj. B</p></li></ul><p><strong>*exclude liquids and solids when calculating</strong></p>](https://knowt-user-attachments.s3.amazonaws.com/3f42dc01-bfed-4509-a865-66657bf7f7ec.png)
Conj. acid base pairs
Conj. acid base pairs differ in the presence or absence of one proton (H+)
Conj. A has one more H+ than conj. B and therefore has a charge that is higher by +1
Relationship between Ka and Kb
(Ka of Ha)* (Hb of A-) = Kw = 1.0 10^ -14
This relationship is true for any conj. Acid-base pair
*This ONLY works for conj. acid-base pairs
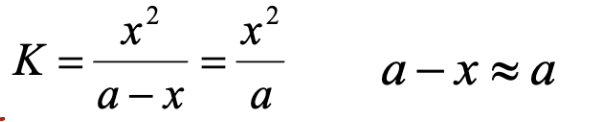
5% rule
When solving for the unknown (x) in an equil. Problem
Where a is the initial concentration of weak base or acid
You can ignore the x in the denominator if doing so gives a % ionization less than 5%
If the % ionization turns out to be greater than 5%, then you can’t ignore x in the denominator and need to resolve w/ the quadratic equation
Titration: can determine [ ] of H2CO3, HCO3-, and CO32- in water
Sample can be titrated with a strong base (like NaOH)
Or strong acid (like HCl)
* has to be strong
* Na and Cl are spectator ions
Which Reaction Arrow to use
If just a weak acid or weak base in water, use ⇌
Ex, acetic acid, ammonia
If one reactant is “strong” (acid or base), use →
Ex. acetic acid w/ NaOH, ammonia with HCl
Acid-base titration
Titrant: solution containing a known concentration of a reactant. Add w/ a burette
Analyte: solution containing a reactant that we want to learn something about (such as molar mass, pKa, pKb, the primary “form” in a solution at certain pH
Equivalence point: a quantity of titrant has been added that is enough to consume all of the analyte, the reaction is complete
Endpoint
the pH at which an indicator changes color, for a titration, an indicator should have an endpoint that coincides w/ the pH at the equivalence point, allows the indicator to correctly indicate that all A or :B has been neutralized
pH indicator
weak acids (HIn) with a special property, acid form of indicator (HIn) is a different color than its conj. Base form (In-), they work well at determining the pH w/in +/- 1 unit of the unit indicator pKa
Limited solubility
compounds that form precipitates, ionic compounds w/ carbonate (CO32-) and silicate (SiO4 4-) most often have limited solubility
A little bit of the compound does dissolve though
Ex Mg(OH)2 has limited solubility and forms a “milky” suspension (not a solution), the limited solubility is what makes it a weak base
Can be described with a chemical equilibrium (exclude solids)
When equil is achieved, solution is saturated w/ ions
Equil. Constant for process = Ksp
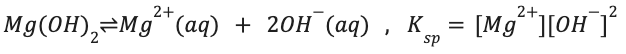
Solubility Equilibria
sometimes we need to increase the solubility of compounds that have limited solubility
Most anions are basic (decent H+ acceptors)
Increase in acidity increases the solubility of the compound
Solubility and precipitate formation
A certain amount of “precipitates” dissolve
If the concentration of dissolved ions are low enough, no precipitate forms
Comparison of the reaction quotient (Q) to the compound’s Ksp value can be used to predict if a solid will be produced in a precipitation reaction
Ksp has a fixed value at a given temp, the pdt of the 2 ion concentrations at equil. Must have this value regardless of how equilibrium is approached, you can start with just reactants, just products, or a mixture of both
*Ksp excludes solids and liquids
Q and K
The reaction quotient (Q) has the same form as the equilibrium constant (K)
For K, the concentrations must be those at equilibrium
For Q, the concentrations can be those at any point in the reaction
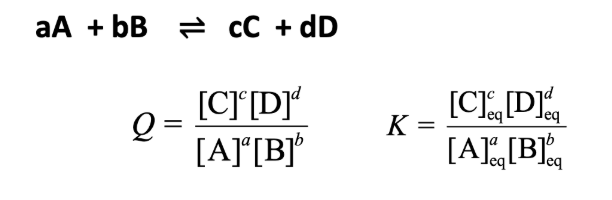
Q vs. K
Q<K → no precipitate, substance will dissolve
Q>K → precipitate
Q=K → at equil, solution is saturated w/ ions an just at the point where if any more is added it will precipitate

Buffers
Near half equivalence points
Solutions which contain appreciable amounts of both a weak acid and its conj. Base ( or a weak base and its conj. Acid )
Resist changes in pH upon addition of a strong acid or base
How they work:
They can neutralize small amounts of added H3O+ and OH- ions
Added H3O+ or OH- are consumed by the buffer and the impact on the pH is minimal
Buffers can be used to maintain a nearly constant pH upon addition of relatively small amounts of strong acid or base
buffers work as they contain existing weak acid or base that soaks up the added acid or base (H3O+/OH-), therefore keeping the pH steady
Common-Ion effect
Definition: the shift in the position of an equilibrium caused by the presence or addition of an ion taking part in the reaction
The ionization of any weak base (A-) is suppressed when a significant amount of its conjugate acid (HA) is already present
The ionization of any weak acid (HA) is suppressed when a significant amount of its conjugate base (A-) is already present
This is an application of Le Chatelier’s principle known as the common-ion effect
The result of this suppression is that the concentration of the species does not change by much
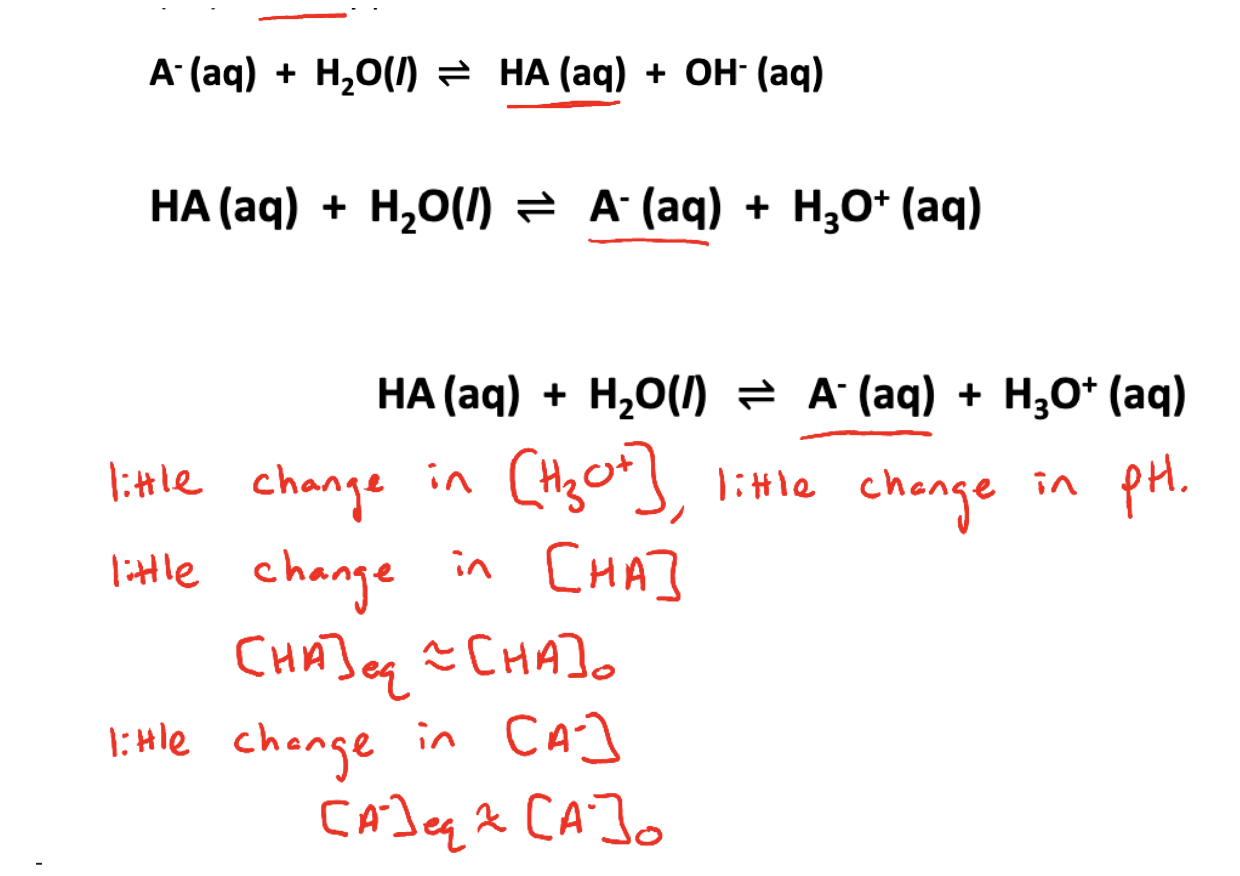
Henderson - hasselbach equation
can be used to calculate the pH of a solution in which the ionization of a weak acid (or base) is suppressed by the presence of a significant amount of its conjugate base (or acid)
Is most accurate when the concentrations of conjugate acid and base are similar (within a factor of 10) → significant amounts of conj. Acid-base pairs
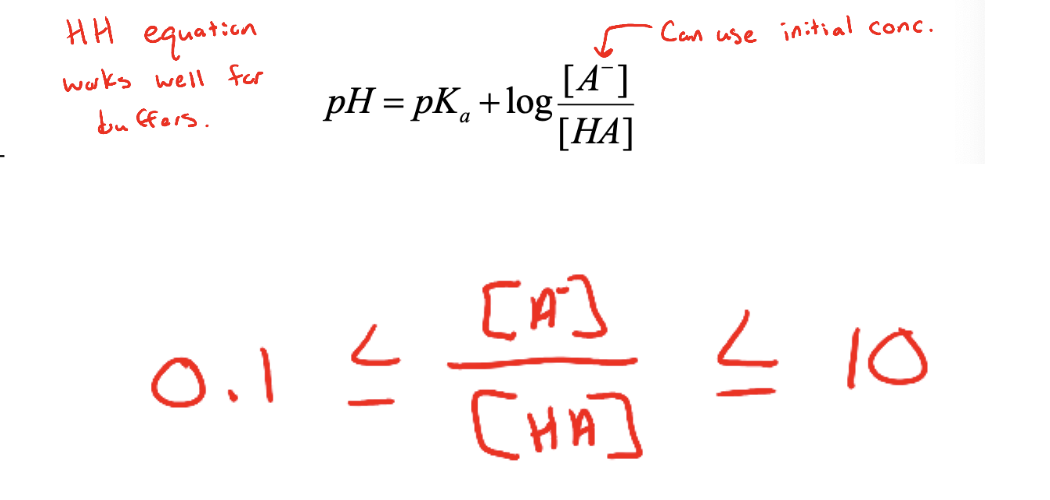
Moles and the Henderson-Hasselbach Equation
If HA and A- are present in the same solution, the ratio of their concentrations is also their mole ratio
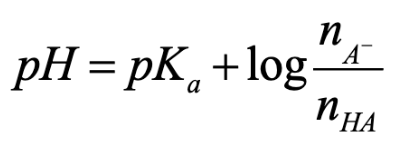
Buffer Capacity
Definition: the quantity of acid or base that a pH buffer can neutralize while keeping its pH within a desired range
all buffers have a limited capacity of how much H3O+ or OH- they can neutralize before large changes in pH take place
Generally, a buffer begins to lose its usefulness if one component is less than 10% of the other

Selection of suitable buffer mixtures
To select an appropriate buffer, one must know the pH range that they want to maintain
From HH equation, we can see that the pH of a buffer depends on 2 factors:
1) pKa of the acid, this has the greatest influence on the buffer pH
2) the ratio of [A-]:[HA]
To obtain a slightly more acidic buffer, add more weak acid → [A-]:[HA] < 1
To obtain a slightly more basic buffer, add more weak base → [A-]:[HA] >1
Ideally want [A-]/[HA] o be between 0.1 and 10:
log(0.1)=-1
log(10)=+1
Buffers work best at pH=pKa +/- 1
Solid Types
Nonmetallic
Ionic
Metallic
Nonmetallic Solids
btwn nonmetals or metalloids
2 types:
Covalent → covalent bonds (much stronger!)
Molecular→ held by IMFs
Ionic Solids
Btwn ions
Brittle, crys. Struct,
When F app, atoms move ∴ atoms w/ like charge touch, ∴ breaks apart
Metallic Solids
btwn metals + metalloids
Malleable, crys. Struct.
Have weak bonds
Electrical conductors
Special type of covalent bonding in metals
M’s share e- w/ mult. Atoms (ex Cu shares 1 e- w/ 12 other atoms)
Allows atoms in adj. Layers to slip past each other
Explains malleability + conductivity
Reg. covalent bonds
share e- pair btwn pair of atoms, σ bond
Allotropes
same element + phys. state, dif. form + properties
Ores
natural compound/mix.
pure elements can be extracted from them
Mostly composed of ionic solids
Crystalline solid
part.’s arranged in 3D array
Slow cool/precip. → atoms have time to get in pos.
Amorphous solids
Lack ordered internal structure
No time for atoms to get into pos.
Malleability
To bend w/ F
M = neutral, no e- repel ∴ lattice don’t break
Hcp + fcc = “smoothest” ∴ “slip” easiest
Metallic bond strength factors
# val. e-
# neighboring atoms
Atom size
↑size, e- further from nuclei, ↓stability, ↓attract. btwn nuclei ∴ weaker bond
*good conductors, sea of e-
P.T. trend for atomic r
↓ group, ← period, ↑r
Packing efficiency
% of total V of unit cell occupied by spheres
2 closest packed: hcp and ccp
Mat.’s of cubic cells tend to form 4-sided crystals
Packing eff. = ((Vatoms)/(Vunit cell))*100
Square packing
atoms in 1st layer (a) touch 4 adj. atoms (most eff. w/ 6)
2nd layer directly above first = cubic packing (simple cubic)
2nd layer nestled in spaces created by (a layer) and pattern continues abab → bcc