Amino Acid Metabolism
1/30
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
31 Terms
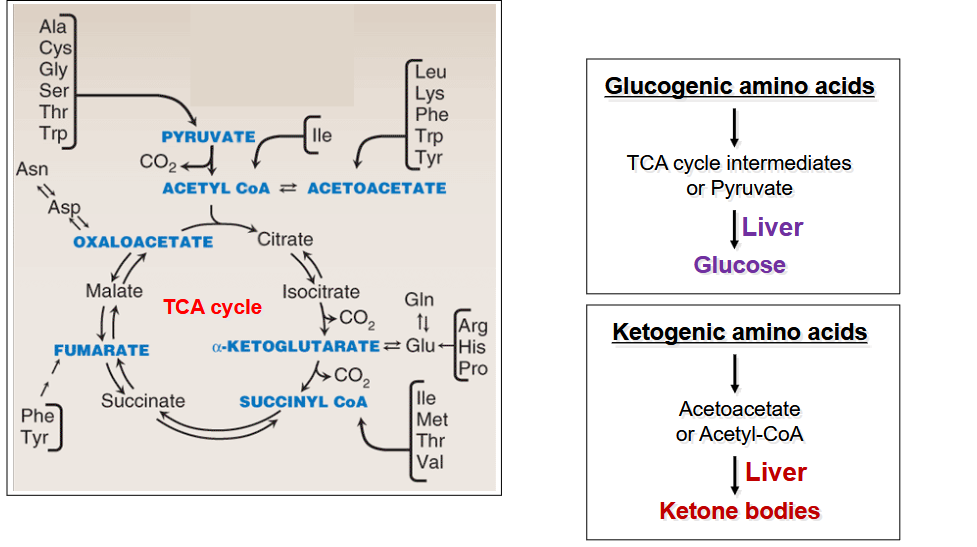
describe the fate of the carbon core of Amino Acids
depends on state of energy
fed state: creation of glycogen and triglycerides
Fasting state: TCA intermediates, pyruvate, acetyl-CoA, acetoacetate, CO2
In both: makes physiologically important metabolites
What organ can degrade all amino acids?; draw out the summary of the degradation of amino acids
liver;
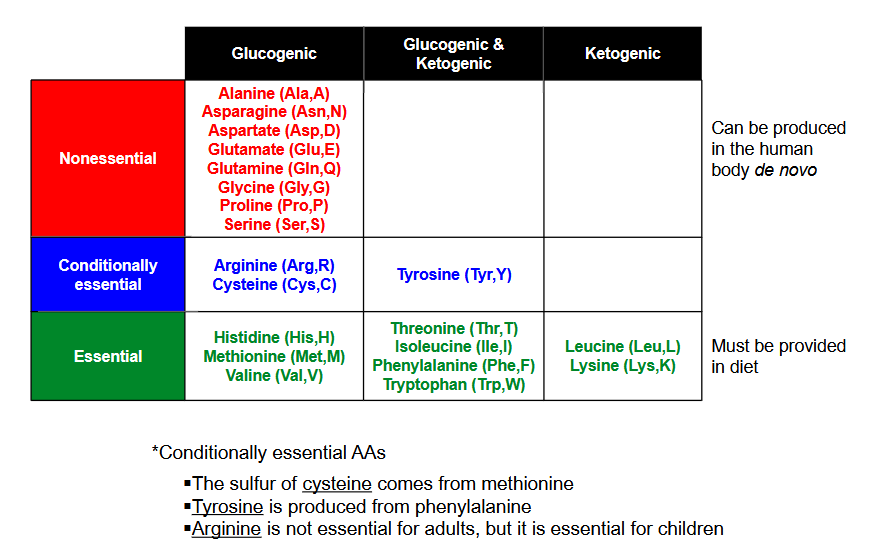
Draw out the chart classifying nonessential, conditionally essential and essential for glugogenic, glucogenic/ketogenic and ketogenic
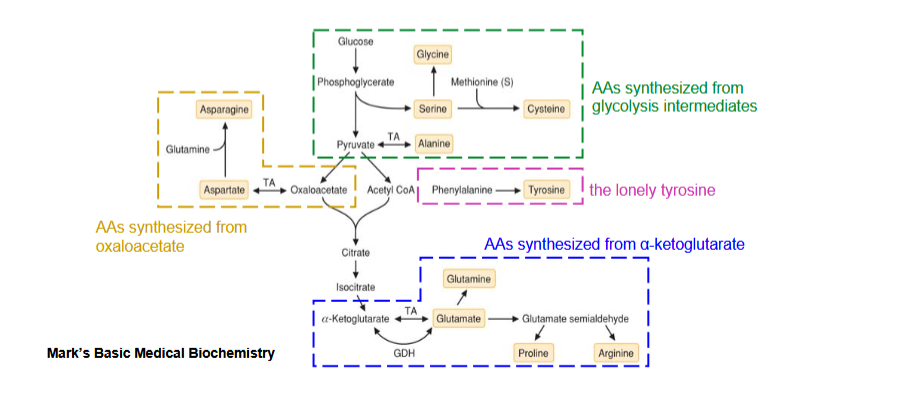
outline the synthesis of nonessential amino acids
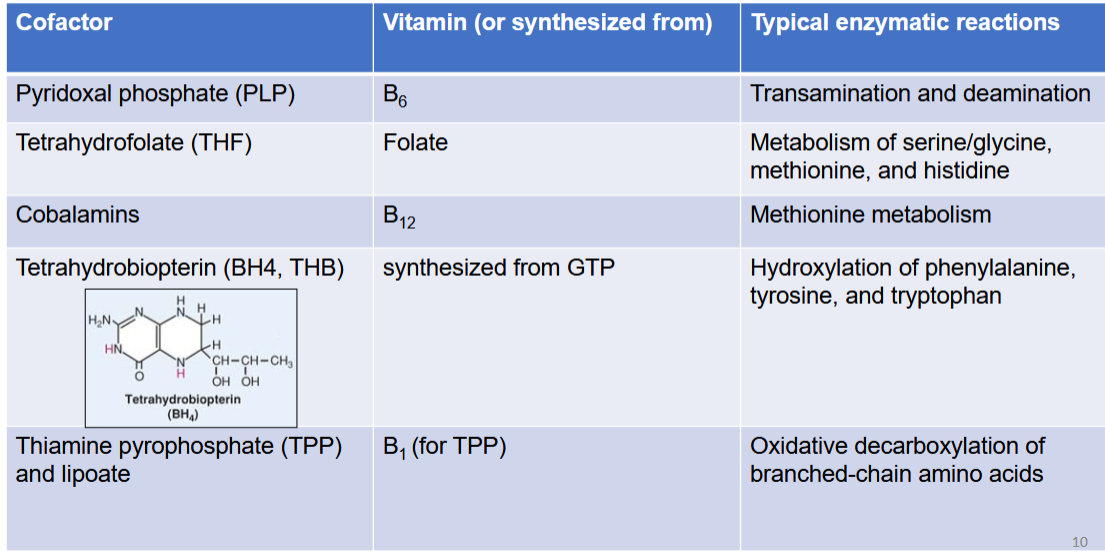
list out the cofactor in AA metabolism, its vitamin, and typical enzymatic reactions in context of AA metabolism
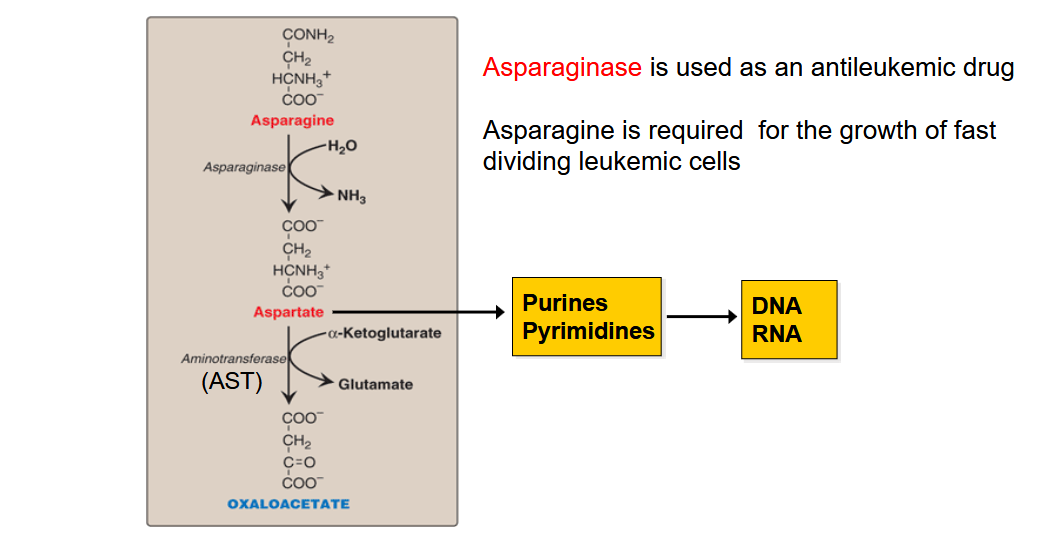
Draw out the pathway for AA that are degraded to OAA; Why is asparaginse use as an antileukemic drug?
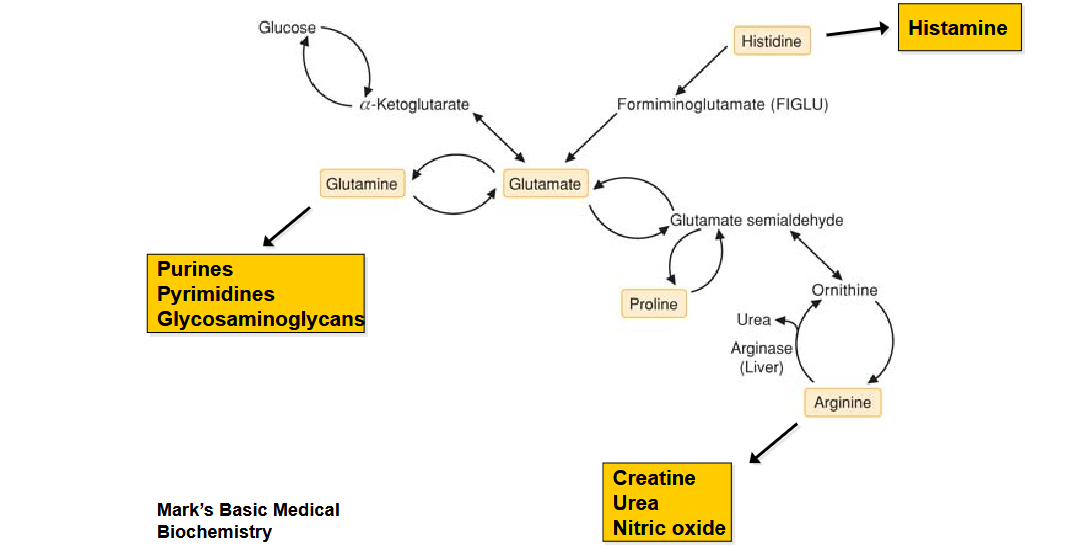
Draw out the pathway for AA that are degraded to a-ketoglutarate
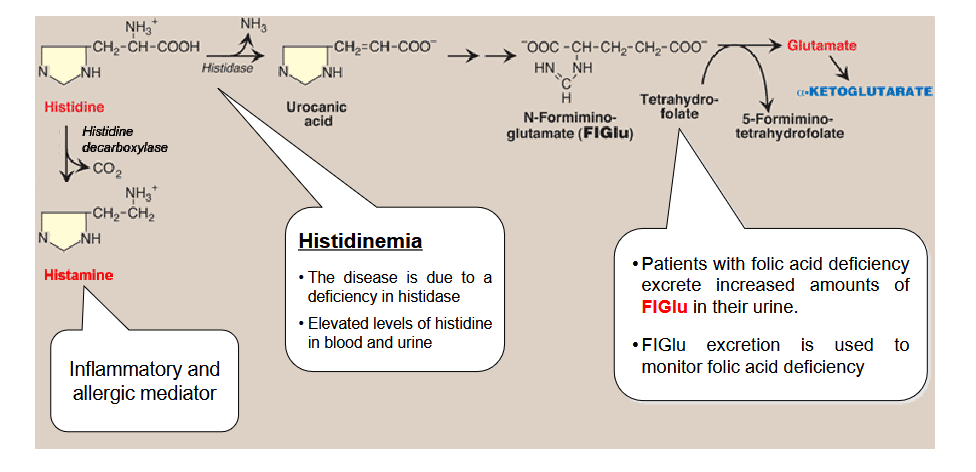
Draw out the pathway for histidine metabolim; What does histamine do? What is histidinemia? What do patients with folic acid deficiency excrete in surplus?
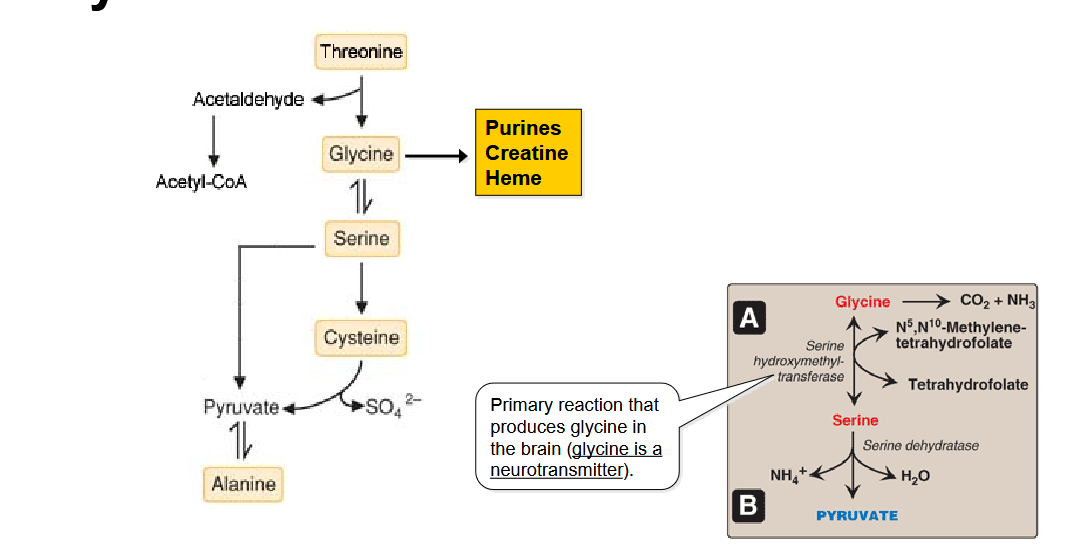
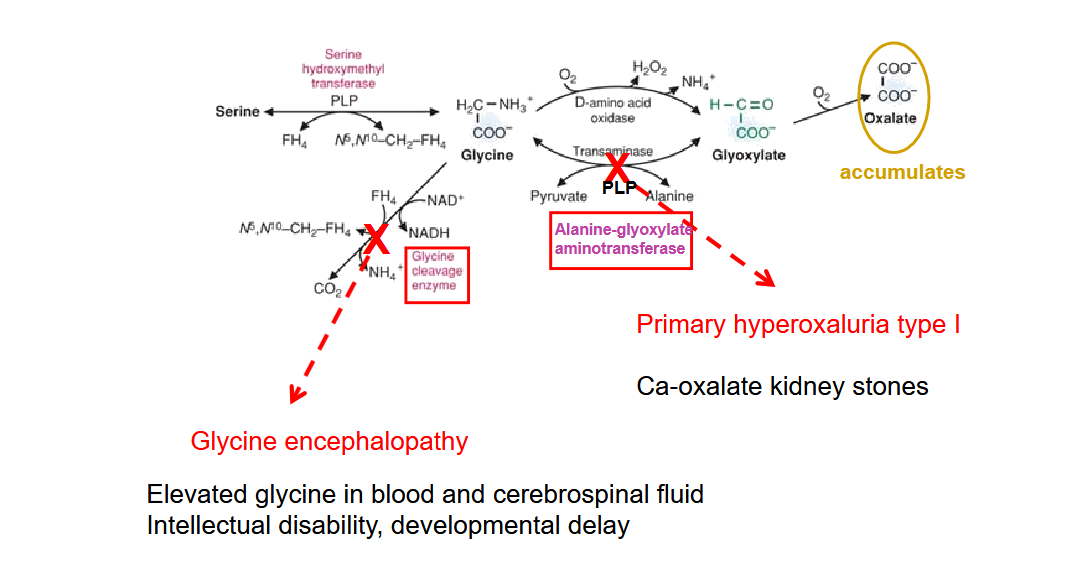
Draw out the pathway for Amino Acids degradation to Pyruvate. What is the primary reaction that produces glycine in the brain?
Draw out the glycine degradation pathway and the two diseases that could arrise from this.
Describe the pathway of making homocystein

What is the primary methyl donor in the body and what does it participate in the syntehsis of?
SAM (s-adenoylmethonine)
participate in:
phosphatidyl choline (cell membrane lipid)
creatine (muscle energy metabolism)
melatonin
epinephrine
draw out the pathway for degradation of homocystein
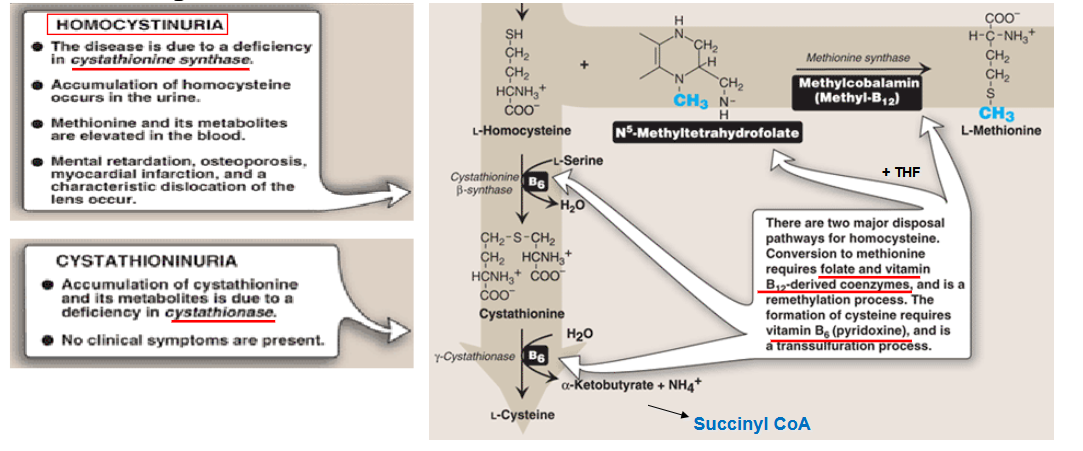
What is homocystinuria? Symptoms?
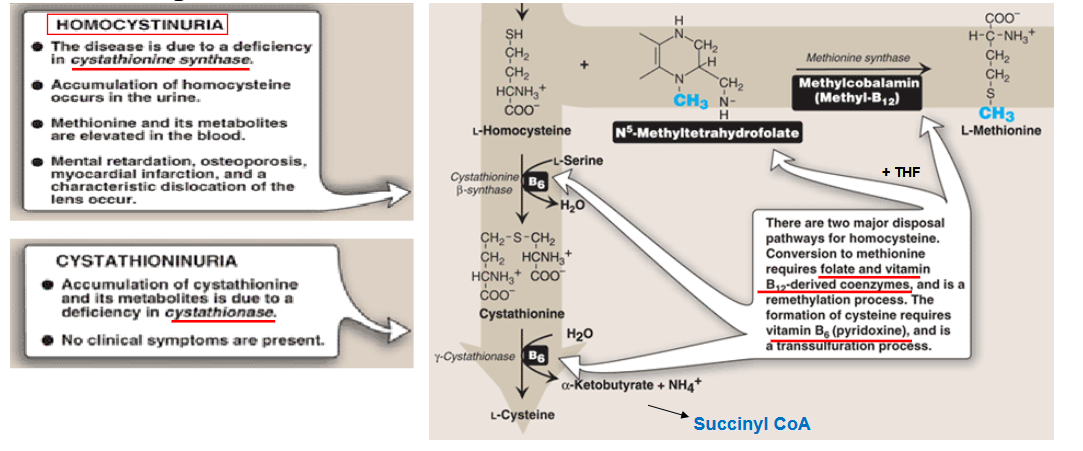
What is cystathioninuria? Symptoms?
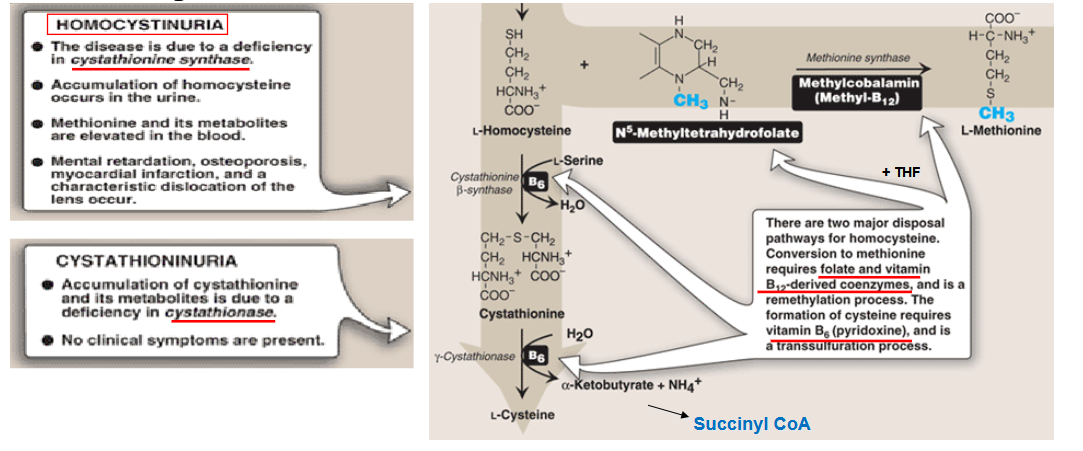
what is high levels of homocystein a risk factor of? How is it treated
high levels of homocystein = risk factor for myocardial infarction; management = restrict methionine, supplement cysteine
draw out the chart describing how cystathionase, cystathionine synthase deficiency and B12 deficiency affect blood levels of methionine, homocystine and cystathionine

What are symptoms and treatment of Homocystinuria?
Symptoms:
elevated homocystine levels in blood/urine
intellectual disability or developmental delay
ectopia lentis (dislocated lens)
skeletal abnormalities (marfan-like)
increased risk of myocarial infarction
treatments:
dietary restriction of Met, and supplementation of Cys
Vitamine B6 supplementation if responsive
Vitamine B12 and folic acid supplementation if responsive
betaine supplementation (methylation of homocysteine)
Draw out the pathway for degradation of Amino Acids to Succinyl CoA
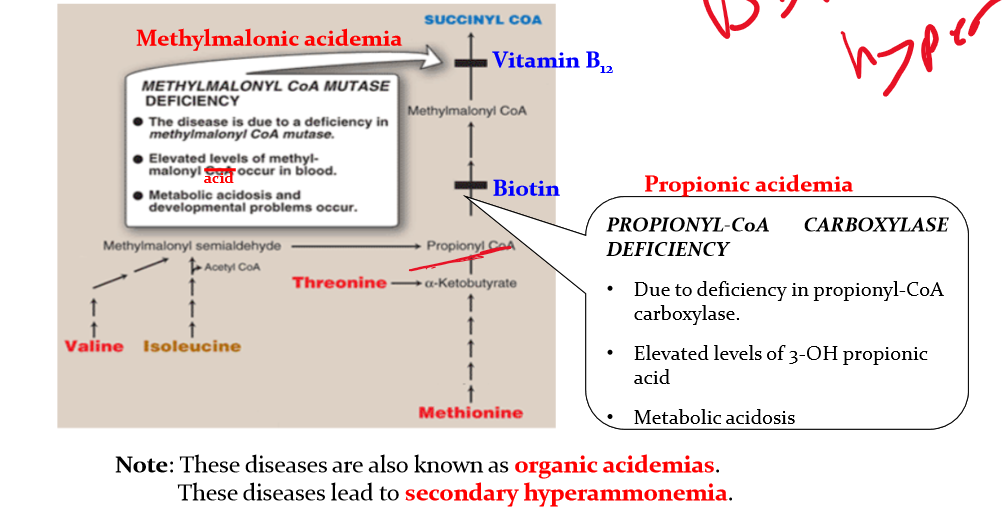
Differentiate between methylmalonic acidemia an propionic acidemia
methylmalonic acidemia:
methylmalonhyl CoA mutase deficiency
elevated levels of methyl-malonyl acid occur in blood
metabolic acidosis and developmental problems occur
Propionic aciemia
due to deficiency in propinyl-CoA carboxylase
elevated levels of 3-OH propionic acid
metabolic acidosis
these diseases are known as organic acidemias and lead to secondary hyperammonemia
draw out the pathway for AA degradation to fumarate
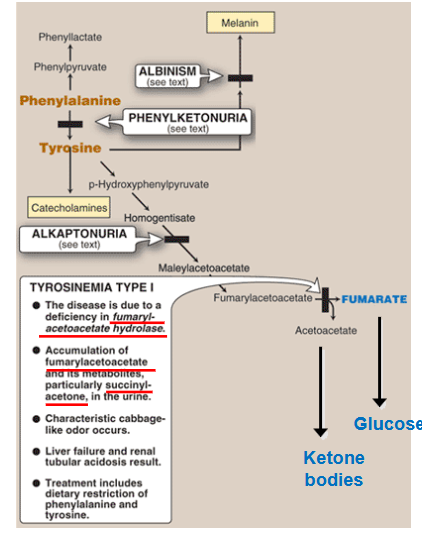
Describe tyrosinemia Type 1
deficiency in fumarylacetoacetate hydrolase
accumulation of fumarylacetoacetate in hepatocyte
hepatic failure, jaundice, cirrhosis, hepatocarcinoma and failure to thrive; associated with very high alph-fetoprotein
accumulation of succinyl-acetone in blood/urine → kidney failure
high excretion of phosphate in urine → rickets (low bone density)
Elevated plasma levels of phenylalanine, tyrosin, and methionine
elevated urine aminolevulinic acid levels
inhibition of heme synthesis and neurological crisess similar to acute intermittent poryphria
change in mental status, abdominal pain, respiratory failure
characteristic cabbage-like odor occurs
liver failure and renal tubular aciosis
treatment: dietary restriction of phenylalanine and tyrosine.
draw out the important metabolites of tyrosine
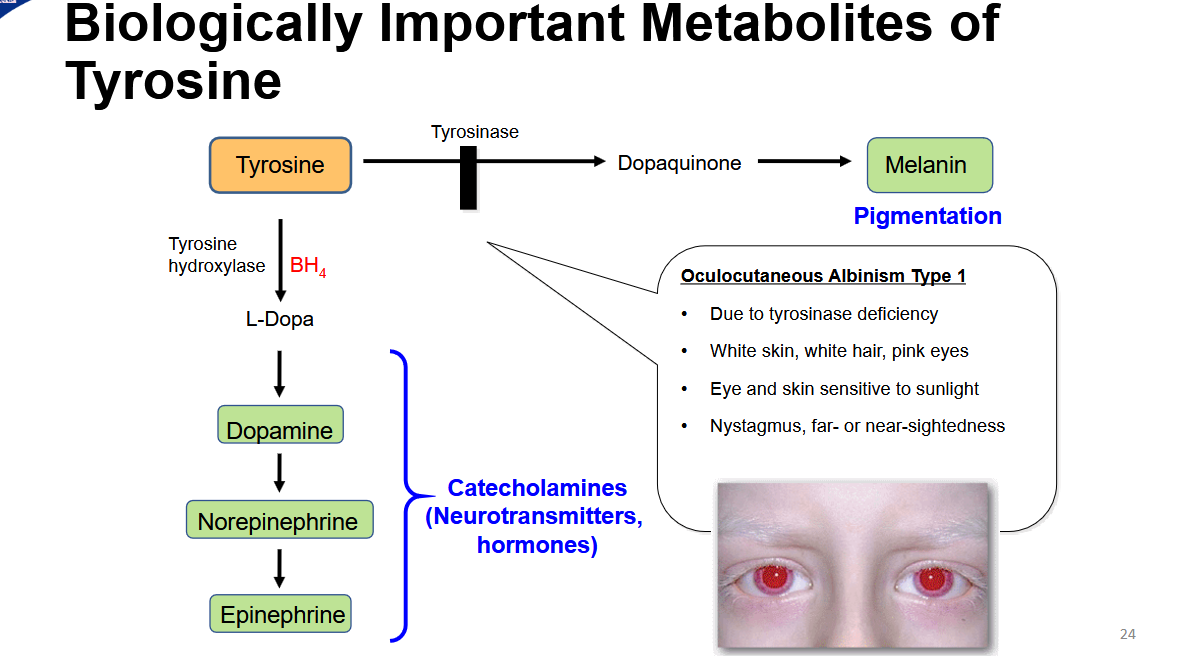
What is oculocutaneous albinism type 1
due to tyrosinase deficiency
white skin, white hair, pink eyes
eye and skin ssensitive to sunlight
nystagmus, far or near sightedness
what is alkaptonuria? discuss this disease’s effect on the urine, eyes, and vertebrae
disease of the tyrosine degraation pathway
due to homogentistic acid oxidase deficiency
homogentistic acid accumulates in urine and tissues
accumulation in cartilate causes crippling arthritis
urine:
after two hours of urine sample, it is entirely black ue to oxiation of homogentisic acid
Eyes:
bluish-black pigmentation in teh sclera of the eye and in teh cartilage o the ear
vertegrae:
dense, black pigment depositied on the intervertebral disk
Describe phenylketonuria (PKU)
due to phenylalanine hydroxylase deficiency
mutation in enzyme or insufficient tetrahydrobiopterin (BH4)
Symptoms:
elevated phenylalanine, phenylpyruvate, phenyllactate, and phenylacetate in blood and urine = musty odor in urine
neurological problems (meental rtardation ,seizurse, tremor, microcephaly, etc) due to reduced production of catecholamins
hypopigmentation due to reduced melanin production
tratment: phe-restricted, tyr-supplemented diet. avoid aspartame, artificial sweetner
neonatal screening is mandatory
What is BH4 necessary in hydroxylation of? why is BH4 deficiency cause mor severe PKU than mutation in phenylalanine hydroxylase?
phenylalanine (tyrosine production)
tyrosine (first step of catecholamine synthesis)
tryptophan (first step of serotonin and melatonin synthesis)
More severe because more pathway inhibited
draw out the pathway for Branched-Chain amino Acid degradation
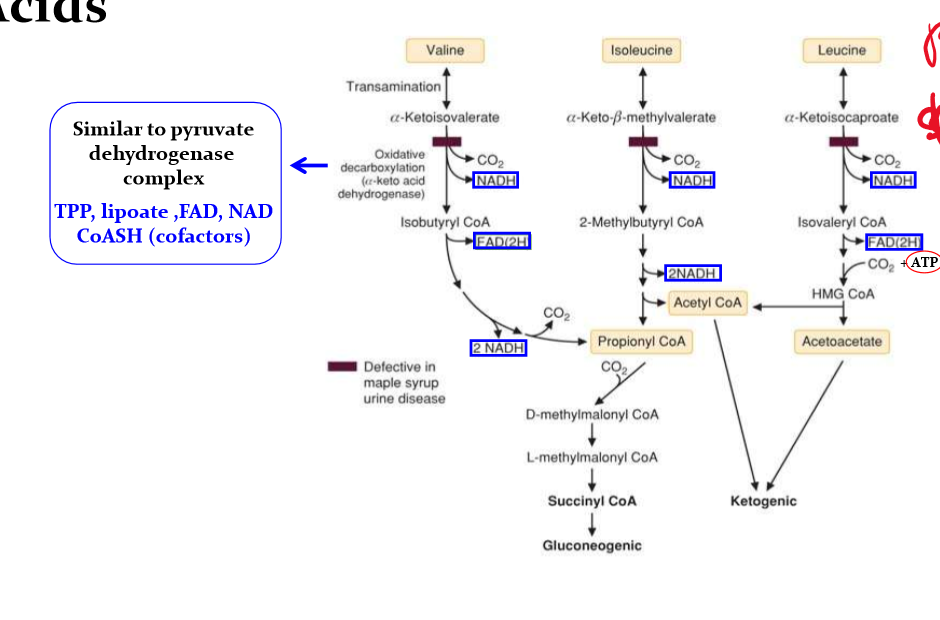
Describe MSUD
Maple Syrup Urine Disease
due to deficient a-keto dehydrognas
Symptoms/lab result
elevated branched chain amino acids (BCAAs) especially leucine in blood
elevated branched chain ketoacids (BCKA) in blood and especially in urine
BCKA causes maple syrup odor of urine and of earwax
encephalopathy (lethargy, poor feeding, apnea, opisthotonus, coma)
Management:
BCAA restricted high calorie diet: usually BCAA-free formulas, supplmented with limited amounts of BCAAs. Leucine is always the most restricted. (all BCAAs are essential amino acid)
draw out the pathway for metabolism of tryptophan
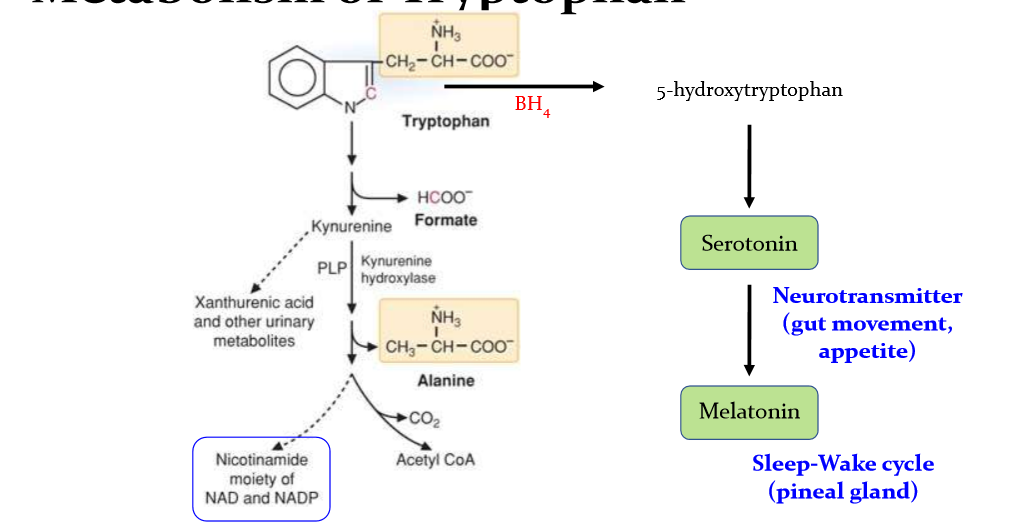
What is pellagra and what are its symptoms?
lack of tryptophan and niacin (vitamin B3) in diet causes pellagra
characterized by 4ds
dermatitis
diarrhea
dementia
death (if untreated)