translational W4 - DNA damage & repair
1/58
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
59 Terms
mechanism of endogenous DNA damage
Damage from within the cell
Internal biochemical processes generate harmful by-products
mechanism of exogenous DNA damage
Damage from outside the cell
Environmental agents that directly interact with DNA
Endogenous Stressors Examples
by-products of cellular reactions interact with DNA
Cellular respiration (ROS) from metabolism (especially from mitochondria)
Lipid peroxidation → creates reactive aldehydes that attack DNA
Spontaneous hydrolysis → can break DNA bonds
Alkylation by endogenous methylating enzymes'
Errors during DNA replication
Exogenous Stressors Examples
Damage caused by environmental exposures:
non-ionising radiation - UV radiation (UV-A, UV-B) → forms pyrimidine dimers
Ionising radiation (X-rays, gamma rays) → causes DNA breaks
Chemicals:
Cigarette smoke, pollution (contain hydrocarbons and alkylating agents)
Aflatoxins (from mould) or other plant/microbial toxins
Chemotherapy drugs (designed to damage DNA in cancer cells)
Diet/lifestyle:
Alcohol consumption
Polyunsaturated fats (can undergo oxidation)
DNA damage → mutation
DNA damage is the first step.
If repair fails, the damage becomes a permanent mutation.
This mutation can be inherited by daughter cells.
DNA damage VS DNA mutation
DAMAGE = reversible, physical or chemical alteration of DNA — Recognised and usually repaired by DNA repair enzymes
MUTATION = irreversible, permanent change in the DNA sequence — Once both strands contain the mutation, not repairable
DNA Damage effect
Can block replication or transcription
DNA mutation effect
May be silent, harmful, or lead to disease or cancer
Some mutations add genetic variation (e.g. SNPs – single nucleotide polymorphisms)
what are SNP’s?
mutations that occur in original sequence over some time
may not have any significant effects or outcomes on specific individuals
change that occurs in small portion of population
what are the two types of DNA mutations
base substitutions (silent, nonsense, missense)
point mutations (transition, transversion)
transition VS transversion point mutations
transition – pyrimidine to pyrimidine OR purine to purine
transversion – pyrimidine to purine OR purine to pyrimidine
which bases are pyrimidines?
C, T, U → 1 carbon nitrogen ring base
which bases are purines?
A + G → 2 carbon nitrogen ring base
what is a silent base substitution?
change in third position of codon, no effect on functional protein
what is a nonsense base substitution?
inserts stop codon, has a non-functional effect - results in truncated protein
what is a missense base substitution?
amino acid change, may change functional protein or may have minimal effects
what is an indel? what is the effect?
what: deletions or insertions within coding region
effect: causes a frameshift mutation – alters translational reading frame
variety of possible outcomes → may cause dysfunctional disease

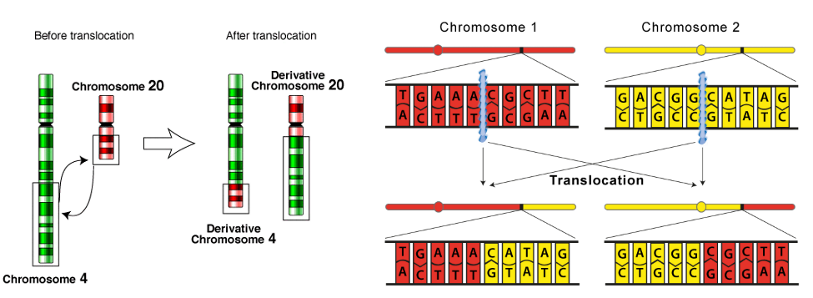
what are DNA translocations and what is the effect?
what: rearrangements between non-homologous regions of chromosomes (e.g. swapping of arms)
outcome: rearrangement of sequence → i.e. promoter shifted = changes regulation of genes by changing their conditions relative other promoter which can have significant functional effects
gene / protein fusions
inactivation
Overexpression
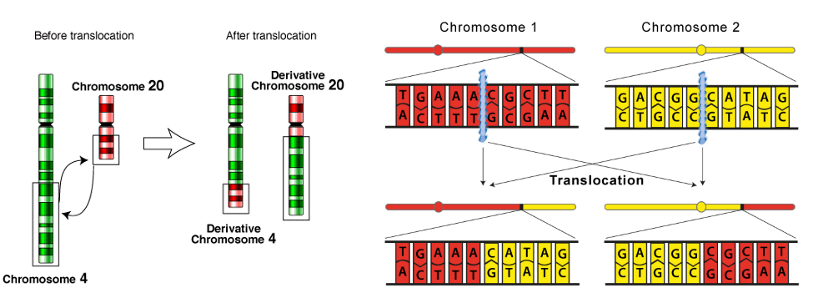
what causes DNA translocations?
DNA double strand breaks
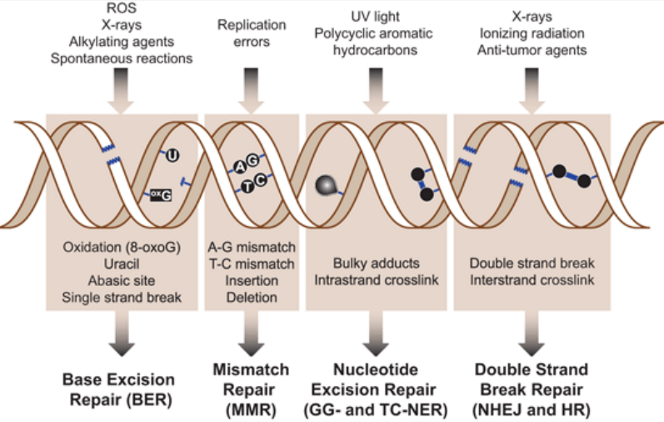
what are the five major DNA repair mechanisms Mammalian cells use;
base excision repair (BER)
mismatch repair (MMR)
nucleotide excision repair (NER)
double-strand break repair
homologous recombination (HR)
non-homologous end joining (NHEJ)
base excision repair (BER)
mismatch repair (MMR)
nucleotide excision repair (NER)
double-strand break repair
homologous recombination (HR)
non-homologous end joining (NHEJ)
What are the 4 classes of DNA repair?
direct repair
excision repair (MMR, BER, NER)
double-stranded break repair (HR, NHEJ)
interstrand crosslink repair
endonucleases function
creation of nicks in DNA to allow access to and removal of damaged region (cleave phosphodiester bond in DNA)
exonucleases function
cleavage of DNA and nucleotide removal (cleave phosphodiester bond at end of DNA)
DNA helicases function
DNA unwinding to allow access of other proteins
DNA polymerases function
addition of new nucleotides
DNA ligases function
stitching together (ligating) the newly created nucleotides to the existing strand
scaffolding proteins function in complex DNA repair
various roles → allow interaction and / or attraction of various repair proteins to sites of damage
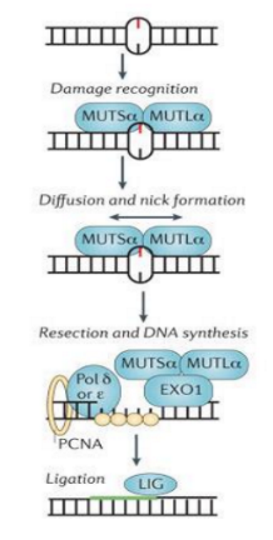
EXCISION REPAIR PATHWAYS - mismatch repair (MMR) corrects:
single base mismatch
insertion / deletion
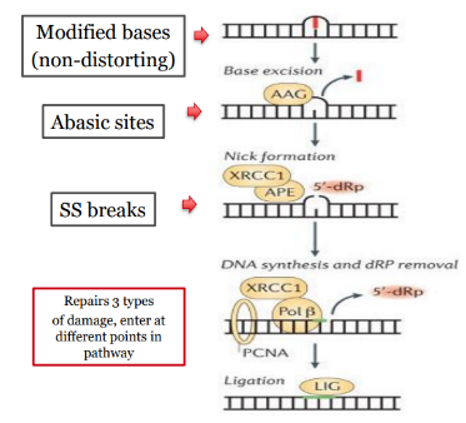
EXCISION REPAIR PATHWAYS - base excision repair (BER) corrects:
small base modifications
abasic sites (AP sites)
single strand (ss) breaks
EXCISION REPAIR PATHWAYS - nucleotide excision repair (NER) corrects:
bulky or helix distorting damage
interstrand crosslinks
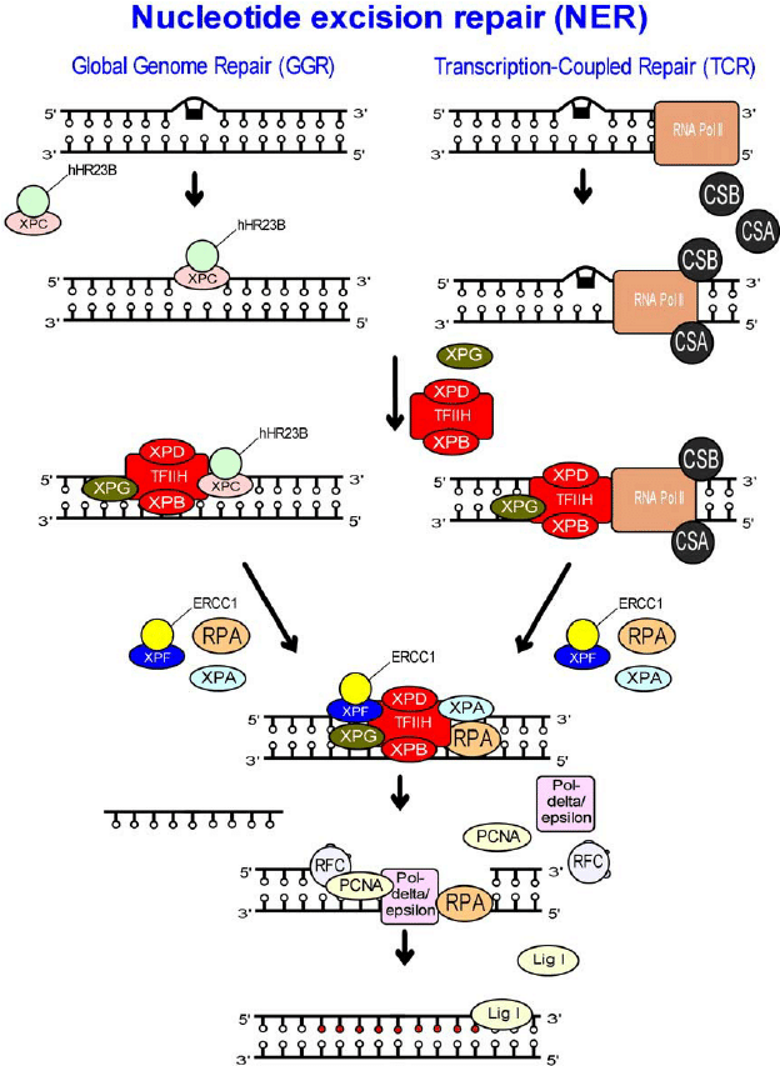
what does GGNER stand for?
global genome nucleotide excision repair
what is the damage recognition molecule in GGNER
XPC-RAD23B
(GGNERR = global genome nucleotide excision repair)
which molecules detect bloackage in Tx-coupled NER?
CSA/CSB
which polymerase is used in translation synthesis as form of DNA repair? What is unique about it?
Translesion polymerase
only inserts 1 - 2 bases
lacks proofreading ability → no endonuclease activity
error prone → often inserts wrong base causing mutation
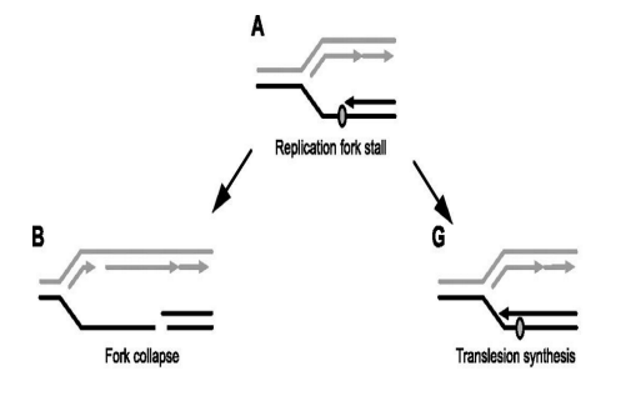
what is the trade-off taking place in Translation Synthesis as a form of DNA repair?
The cell is choosing a small risk (mutation) over a major risk (cell death from fork collapse)
Benefit: avoids catastrophic DNA breaks and keeps replication going.
Drawback: introduces base substitution mutations, increasing the chance of genetic errors.
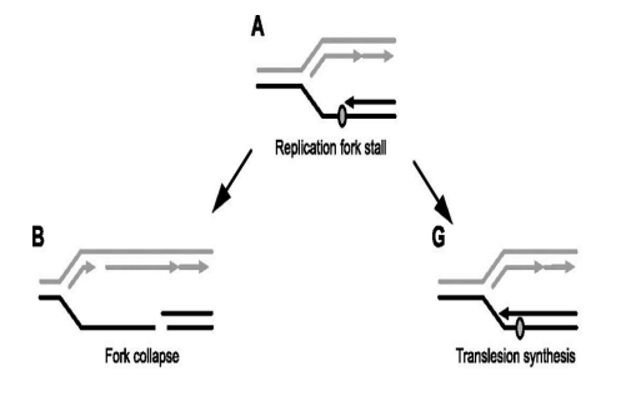
consequences of unrepaired DSB
genomic instability
mutations
cell death
cancer.
Which sensor proteins recognise the ends of DSB and bind the DNA strand?
MRN and Ku70/Ku80 complexes
possible consequences of NHEJ
translocation of lost nucleotides on a damaged DNA strand
loss of nucleotides at the site of repair
interuption of gene expression
which DNA repair mechanism is primarily responsible for repairing DSB?
Homologous Recombination (HR)
error free strategy
can only occur S1/ S2 phase → requires sister chromatic
distinguish between NHEJ & HR
Feature | NHEJ | HR |
|---|---|---|
Template required | ❌ No | ✅ Yes (sister chromatid) |
Fidelity | ❌ Error-prone (small deletions) | ✅ High-fidelity |
Cell cycle phase | All phases (G1, S, G2, M) | Only in S and G2 (requires a sister chromatid) |
Speed | ✅ Fast | ❌ Slower |
Common use | Quick fix, emergency repair | Clean, accurate repair of complex/ ‘unclean’ damage |
Major risk | Translocations, mutations | Limited availability (needs template) |
when is HR or NHEJ preferentially chosen?
HR = accuracy is vital and a template is available
NHEJ = speed is the priority.
What is Artemis role in NHEJ?
trims or modifies the DNA ends
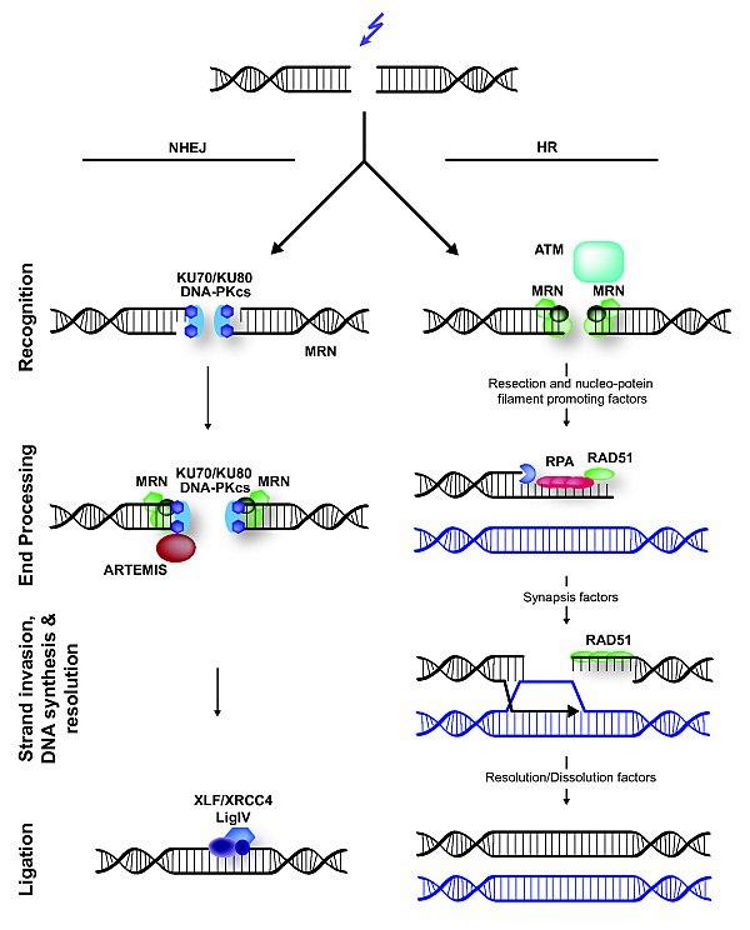
which 2 molecules ligate the ends in NHEJ?
DNA ligase + XRCC4
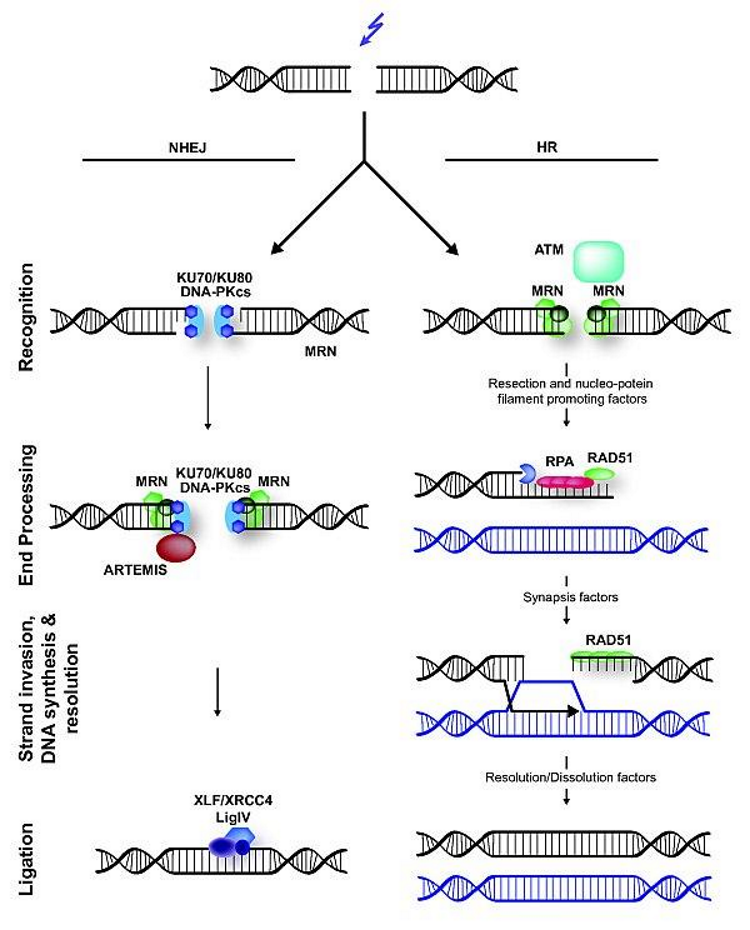
role of RPA in DNA repair
used for Tx-coupled NER & HR
coats the ssDNA
Prevents the ssDNA from forming loops or being degraded
What is the DDR and what is its function??
WHAT: a network of pathways the cell uses to detect, signal, and repair DNA damage.
FUNCTION: It ensures genomic stability, and when damage is too severe, it can trigger cell cycle arrest, senescence, or apoptosis.
Key concept:
Damage → Sensor Proteins → Kinases → Cell Cycle Arrest → Repair OR Death
DDR - Single-Stranded DNA Damage (SSBs and replication fork stalling)
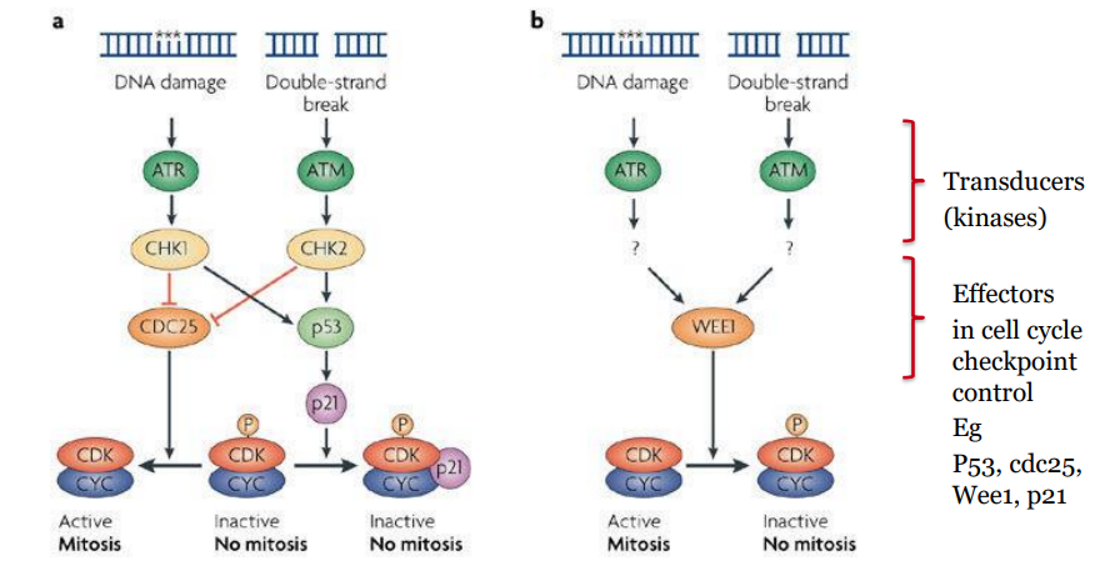
STRETCHES OF ssDNA SIGNAL DDR VIA ATR/CHK1 PATHWAY
DNA replication stalls due to damage (e.g. a DNA lesion).
Helicase continues, exposing single-stranded DNA (ssDNA).
RPA binds to exposed ssDNA → recruits ATRIP
ATRIP activates ATR kinase
ATR phosphorylates CHK1 → inactivates CDC25 phosphatases
CDC25 inactivation → CDK2 + Cyclin E can't function → intra-S phase arrest
SSB or replication fork stalling → RPA → ATRIP → ATR → CHK1 → CDC25 inactivated → S-phase arrest
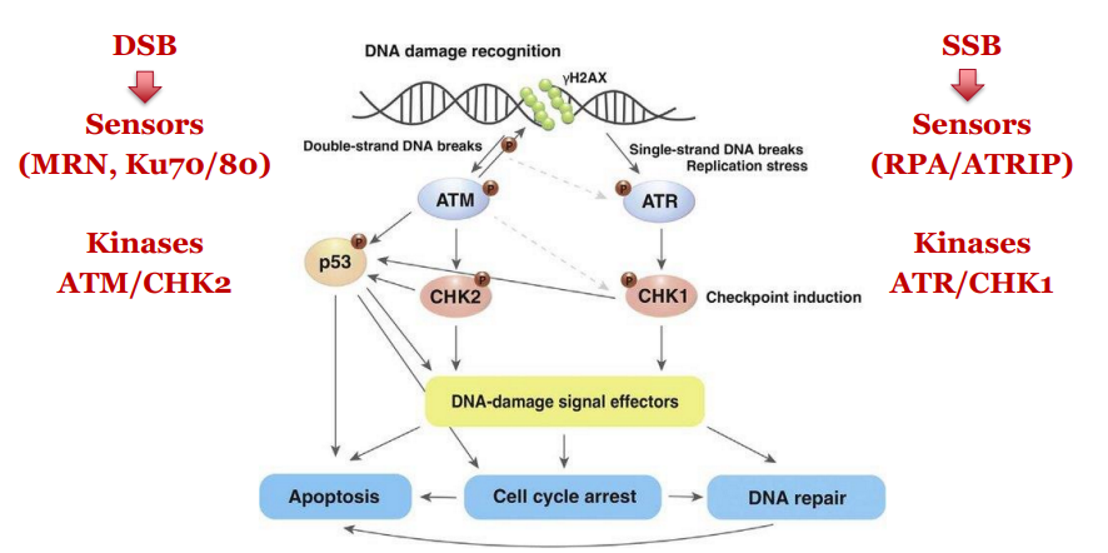
DDR - Double-Stranded DNA Breaks (DSBs)
DSB is sensed by MRN complex and Ku70/80
Activates ATM kinase
ATM phosphorylates CHK2
CHK2 inactivates CDC25 phosphatases → blocks CDK activity → cell cycle arrest
DSB → MRN + Ku70/80 → ATM → CHK2 → CDC25 inactivated → cell cycle arrest
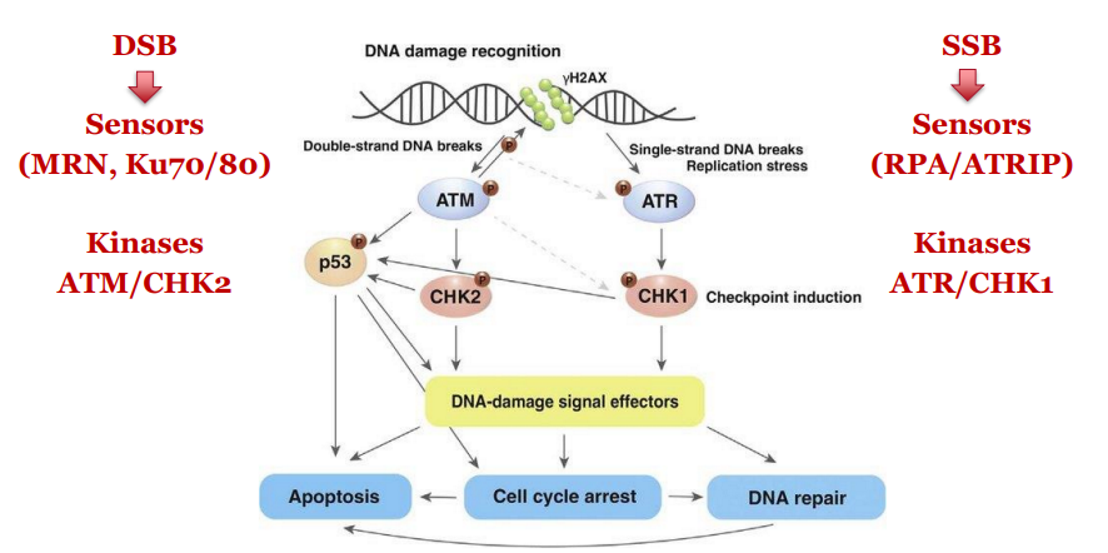
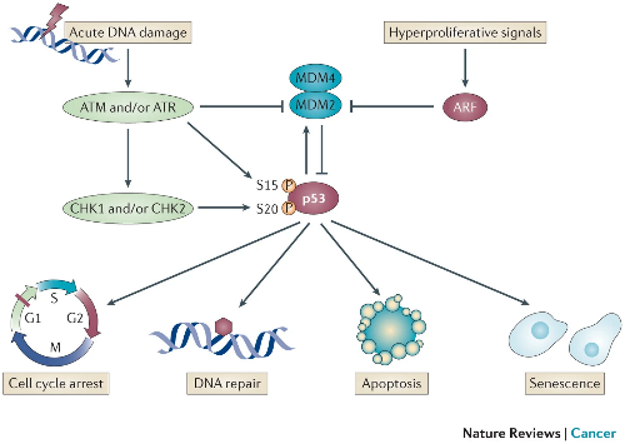
Role of p53 in DDR
p53 is a tumour suppressor Tx factor protein that integrates upstream damage signals to the downstream pathways
activated by phosphorylation of ATM / ATR / CHK1 and CHK2.
When phosphorylated → stabilises protein so p53 avoids degradation (not targeted by MDM2 = Ub ligase).
stabilised p53 increases transcription of p21, which inhibits CDK2.
This reinforces cell cycle arrest and gives time for repair or triggers apoptosis.
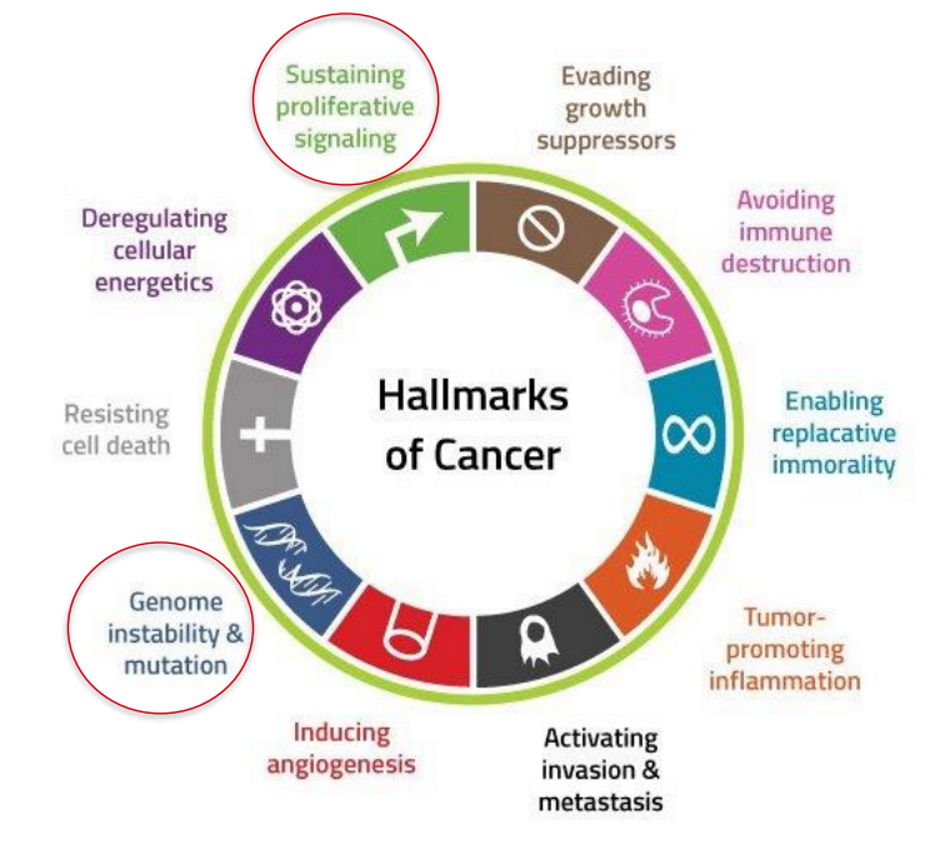
What are the hallmarks of cancer?
early cancerous lesion – uncontrolled growth
tumour progression = step-wise acquisition of various characteristics (hallmarks)
This is acquired by early genome instability
This instability attributed the cell adaptability to complex tumour environments
basic idea of DDR and Cancer
If DDR fails, DNA damage accumulates → genome instability → mutations in key genes (e.g. oncogenes and tumour suppressors) → cancer.
What is the multi-hit hypothesis in relation to Cancer?
Key idea = Cancer develops from the accumulation of multiple mutations over time, not just one.
EXPLAINS: Why cancer needs several mutations
These mutations can affect tumour suppressors, oncogenes, DNA repair genes, etc.
Each “hit” gives the cell a growth or survival advantage.
Often visualised as stepwise progression (e.g. from normal → hyperplasia → dysplasia → carcinoma).
I
What is the Mutator-phenotype hypothesis in relation to Cancer?
KEY IDEA: 1st mutation in DNA repair genes (e.g. MMR, NER) → increases overall mutation rate → step wise accumulation of mutations → higher risk of cancer (accelerates the multi-hit process)
EXPLAINS: How some cells get lots of mutations quickly
Leads to genomic instability early in cancer development.
As they accumulate these mutations they grow faster, survive longer and become 'fitter'
Attributes selective advantage that enables them to evade checkpoints
As they grow have ongoing adaptation = no blood supply, O2, nutrients
Examples of Inherited Disorders with Increased Cancer Risk (supports mutator hypothesis)
Xeroderma pigmentosum → NER defect → XP gene defect → extreme UV sensitivity → reduced capacity to repair UV damage (pyrimidine dimers) → increased risk UV-induced skin cancers
Lynch syndrome (HNPCC) → MMR defect → MutS/L gene prone mutation → ↓ DNA repair → ↑ mutation rate → ↑ risk of additional “hits” →colorectal & uterine cancers
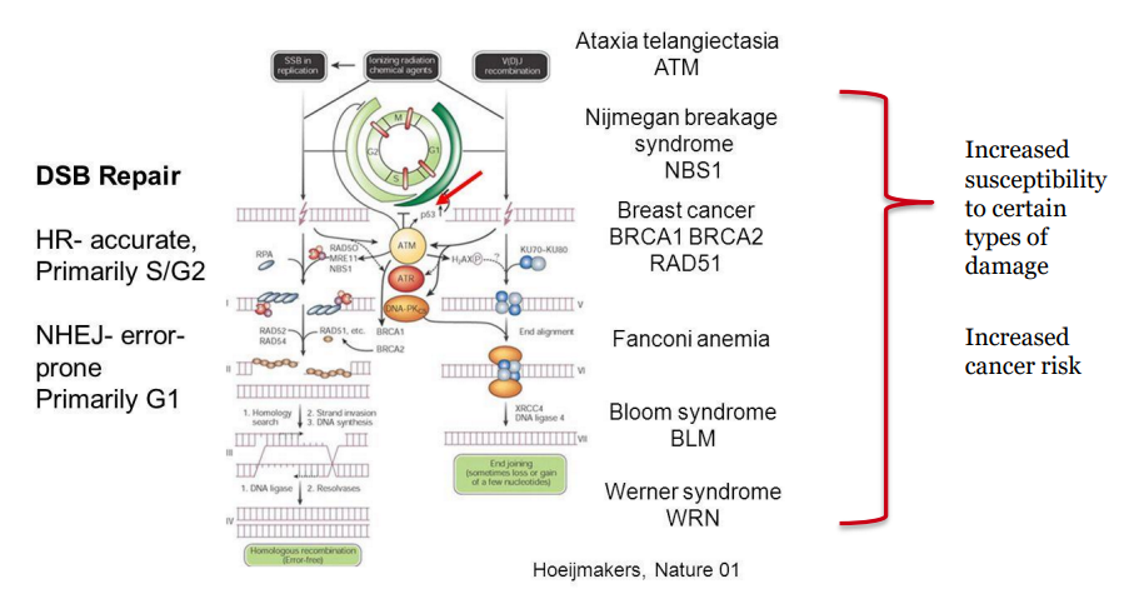
what do the Multi-hit and Mutator Phenotype Hypothesis explain?
Multi-Hit Hypothesis = Why cancer needs several mutations → focus on any gene (oncogene, suppressor, etc.)
Mutator Phenotype = How some cells get lots of mutations quickly → focus on DNA repair genes
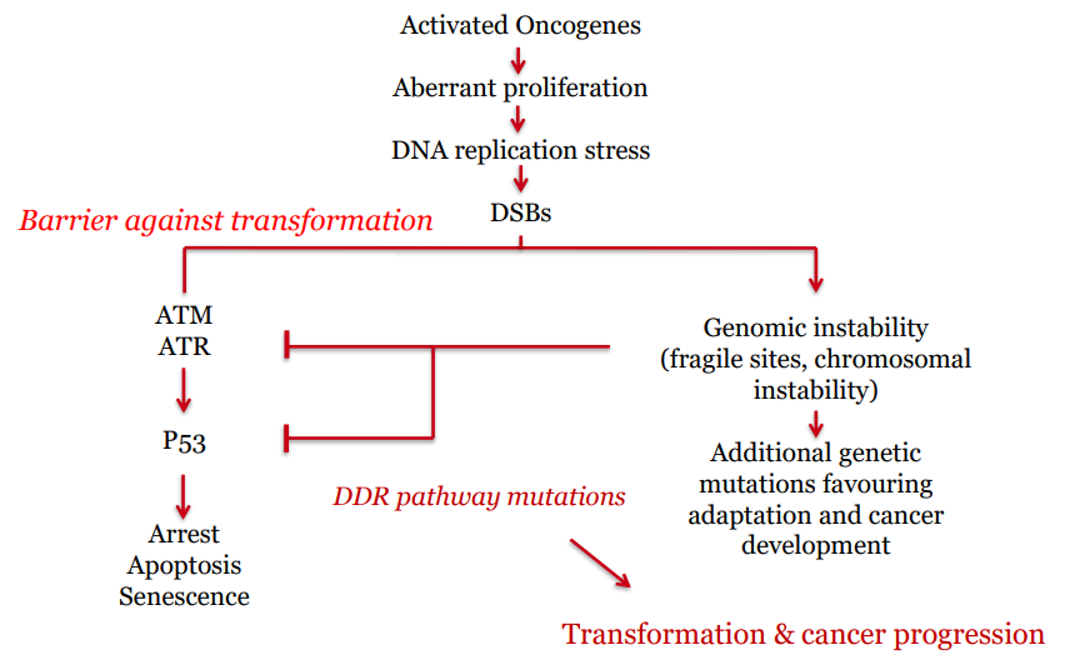
what causes genomic instability in sporadic (somatic) cancers?
Oncogene activation → replicative stress → DSBs → DDR responds
But eventually: mutation in DDR → genomic chaos → cancer progression accelerates
In sporadic cancers, the first mutations are often in proto-oncogenes → turned into oncogenes
This leads to:
Increased cell division
Replication stress (too much or faulty DNA replication)
Resulting in DSBs and DNA damage
more replication = more DNA damage = mutation in DDR genes = uncontrolled proliferation
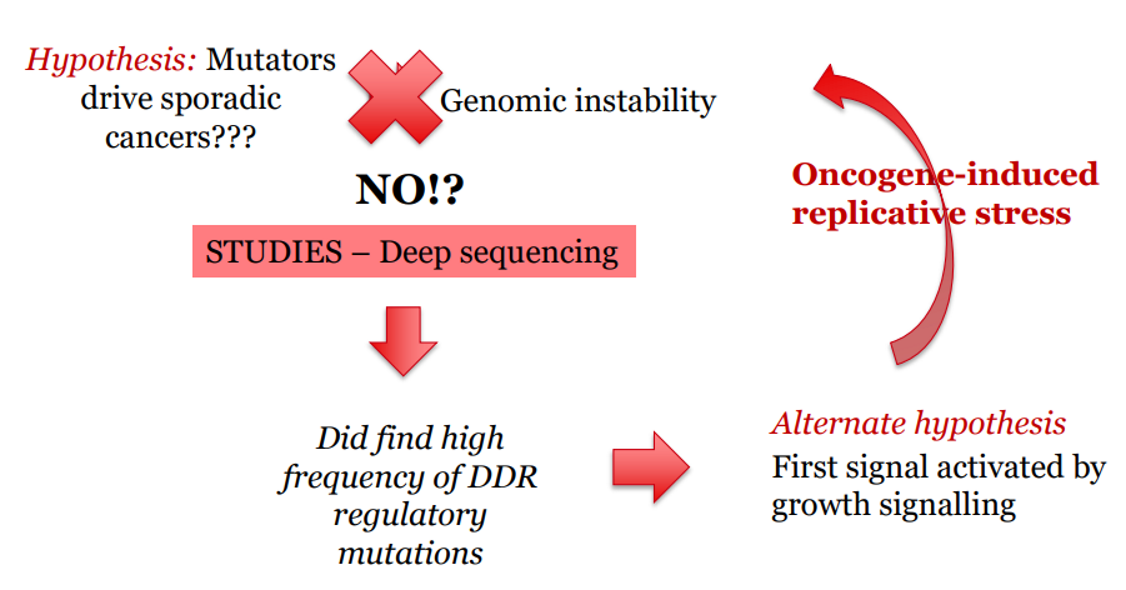
damage – stalled replication forks
oncogenes – alterations to replication timing and progression
If have oncogenes that are just driving cell cycle forward without pausing or looking for damage = exposure of various fragments that are prone to being damaged = double strand breaks
Do oncogene induced tumour progression require the up or down regulation of the DDR?
Down-regulation of damage surveillance mechanisms (DDR).
oncogenes drive excessive proliferation → replicative stress
normally DDR kicks in to kill cell, but cancer cell wants to keep growing
therefore disable/ downregulate so cell can continue dividing
Key Difference: What activates ATM vs ATR?
ATM | ATR |
|---|
Activated by double-strand breaks (DSBs) | Activated by single-stranded DNA (ssDNA) regions, especially at stalled replication forks |
Sensor complex: MRN complex | Sensor: RPA-coated ssDNA recruits ATRIP |
Major downstream target: CHK2 | Major downstream target: CHK1 |
Cell Cycle Effect: Inhibits CDC25 → G1/S and G2/M arrest | Cell Cycle Effect: Inhibits CDC25 → Intra-S and G2/M arrest |
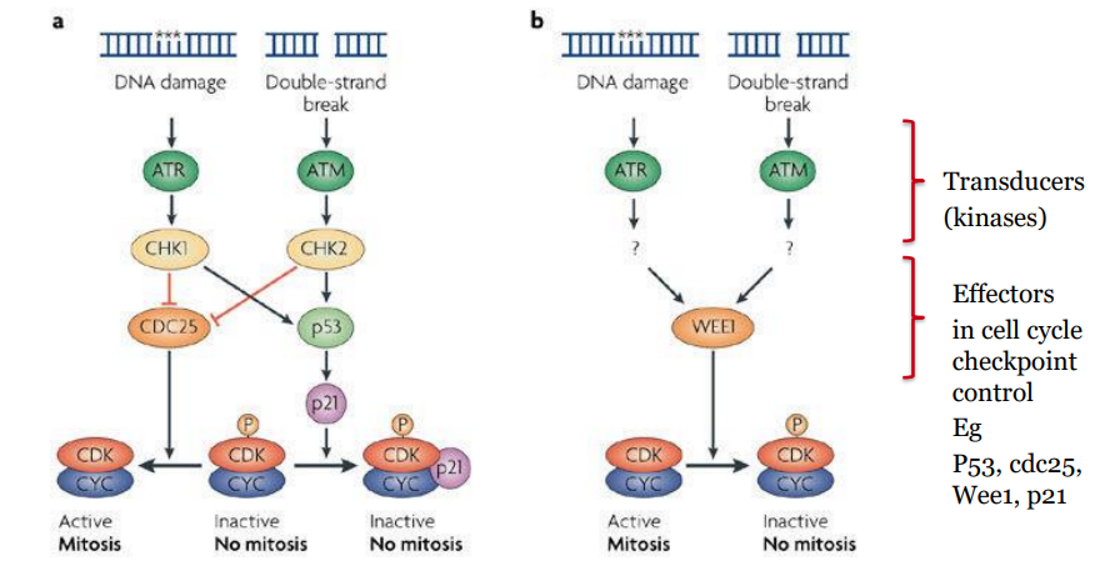
main cell cycle effect of P53
stabilised by CHK1/2 → increases p21 expression → inhibits CDK → reinforces G1 and G2 checkpoints