Flipped Lesson 3: Haemoglobinopathies & Mutation
1/61
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
62 Terms
What does the H locus on chromosome 19 encode?
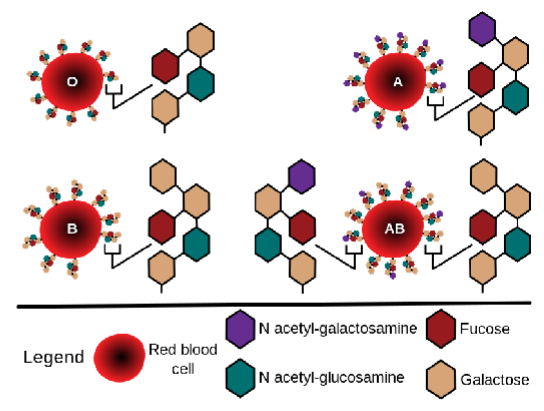
Fucosyltransferase → synthesises the H antigen - responsible for Synthesis of a Sequence of Monomers (Saccharides & Related) on RBC Surface Molecules
What determines the ABO blood group?
The ABO locus on chromosome 9, encoding glycosyltransferase enzymes.
What is codominance
When both alleles of a gene are fully expressed in heterozygotes, producing an additional phenotype (e.g., AB blood group).
How does codominance differ from incomplete dominance?
Incomplete dominance produces a blended phenotype, whereas codominance produces an additional, distinct phenotype.
Draw the specific structure of the O, A, B, and AB groups
What is the composition of normal adult haemoglobin A (HbA)?
Two α-globins (141 aa)
Two β-globins (146 aa)
(written (αβ)₂.)
Haemoglobin genes have Biallelic Expression. What does this mean?
Both maternal and paternal alleles are expressed simultaneously.
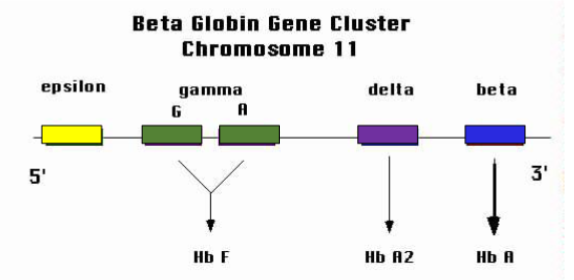
Where is the β-globin gene cluster located & how is it arranged
On chromosome 11, arranged 5′ → 3′ as: ε (epsilon), Gγ (gamma G), Aγ (gamma A), δ (delta), β (beta).
Which haemoglobins are formed from the β-cluster genes?
ε → embryonic Hb (e.g., Hb Gower).
γ (G, A) → foetal Hb (HbF, α₂γ₂).
δ → HbA₂ (α₂δ₂).
β → HbA (α₂β₂).
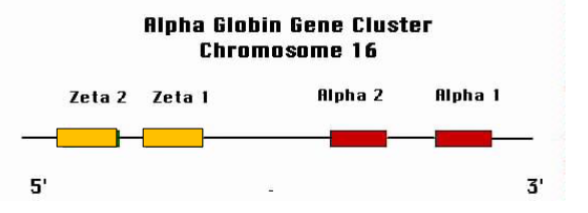
Where is the α-globin gene cluster located & how is it organised
On chromosome 16, arranged 5′ → 3′ as: (zeta 2), (zeta 1), α2, α1
What are the three main sites of erythropoiesis during development?
Yolk sac (very early, weeks 3–10).
Liver (and spleen) (main site from weeks 6–30).
Bone marrow (takes over from ~30 weeks onward and throughout life).
Which globin chains are expressed in the earliest embryonic period?
(zeta) and ε (epsilon).
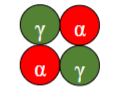
What is the predominant haemoglobin in the foetus?
HbF (α₂γ₂), due to high γ-globin expression.
How does the proportion of γ-globin change after birth?
It falls rapidly after birth, replaced by β-globin production.
When does β-globin synthesis begin to rise significantly?
Around 30 weeks gestation, becoming dominant after birth.
What happens to α-globin expression over development?
It rises early and then remains consistently high throughout life.
What is the physiological significance of HbF predominance before birth?
HbF has a higher affinity for oxygen than HbA, allowing efficient oxygen transfer from maternal blood.
What is the subunit composition of HbF(/HbFF)? & what is it
(α₂γ₂) – foetal haemoglobin.
What is the subunit composition of HbA(/HbAA) & what is it ?
(α₂β₂) – adult haemoglobin.
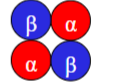
What is the subunit composition of HbA₂(/HbA₂A₂) & what is it?
(α₂δ₂).
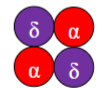
What is the subunit composition of HbS(/HbSS) & what is it?
(α₂βˢ₂) – sickle haemoglobin.
Why is haemoglobin packaged inside RBCs?
Free Hb is rapidly catabolised and excreted by kidneys.
Concentration of Hb in RBCs
320-350g/L of cytoplasm
Close to limit of solubility of Hb in physiological solution
What happens if Hb concentration exceeds solubility limit
Polymerisation → precipitation → distorted RBCs → haemolysis (RBC lysis)
Are individual globin chains (monomers) soluble?
No, monomers are insoluble, but the tetramer is highly soluble.
What in Hb biosynthesis is critical for proper Hb function
Balanced production of α and β chains (Requires co-ordinated Gene Expression)
How is excess α chain managed?
Protease degradation (safety valve), though it can be overloaded.
How is Hb gene expression coordinated?
Chromatin restructuring regulates transcription.
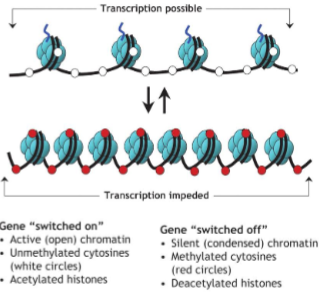
How does the structure of chromatin & genes change when switched on/off
Where are the Hb genes located?
Chr 11 and Chr 16 (these are the locus control regions)
What is the role of the LCR (locus control regions)?
Cis-acting transcriptional regulation of β-globin gene cluster, acting on the same DNA molecule. Do Not Encode for Peptides - they are promoters/enhancers/silencers
cis-acting meaning
acting from the same molecule
intra-molecular action
Act on the DNA strand on which they are encoded
How does transcription & chromatin remodelling change from foetal life → adult life
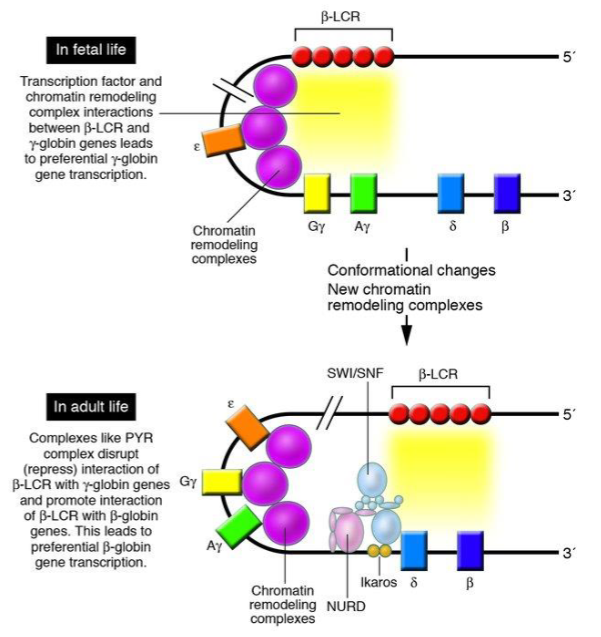
What are the two main types of inherited Hb disorders?
Haemoglobinopathies → structurally abnormal globins (normal amounts, wrong sequence). Causes globin chain polymerisation and misshapen RBCs, e.g. sickle cell
Thalassaemias → imbalance of otherwise normal globin chains. Causes not enough Hb (anaemia) and/or abnormal accumulation of globin subunits (toxic) (β-thalassaemia)
What mutation causes sickle cell anaemia?
Missense mutation in β-globin gene → HbS.
What happens under low oxygen tension?
HbS polymerises → RBCs sickle → obstruct vessels, haemolysis.
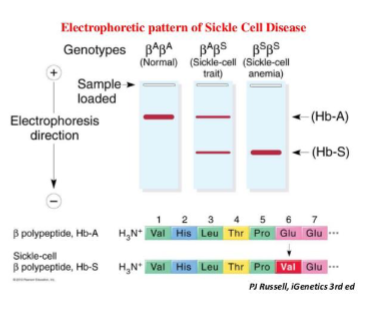
What base substitution occurs in sickle cell disease?
Glutamic acid (GAG) → Valine (GTG) at codon 6 of β-globin.
CTGAGGAGA (HβA gene) – Beta haemoglobin
CTGTGGAGA (HβS gene) – Sickle cell haemoglobin
MIM ID of sickle cell
#603903
Sickle cell Gene map locus
11p15.5
What are the genotypes and phenotypes?
HbAS: heterozygote, sickle cell trait.
HbSS: homozygote, sickle cell anaemia.
How is sickle cell disease diagnosed?
Haemoglobin electrophoresis: Hb variants migrate differently due to charge differences.
Most Common Genetic Disorders of Hb
Thalassaemias
What causes thalassaemias
Defects in balanced biosynthesis of globin chains.
What are the clinical results of thalassaemias
1. Not enough Hb (anaemia)
2. Abnormal accumulation of Globin subunits
What are the two main clinical types of thalassaemias
β – thalassaemia: Absent or reduced β globin
α-thalassaemia:Absent or reduced α globin
What gene is affected in β-thalassaemia and where is it located?
HBB gene on chromosome 11.
2 types of β-thalassaemia
Minor: (heterozygous mutation – one defective gene copy)
Major: (homozygous/compound heterozygous mutations – both gene copies defective
At what stages can mutations occur?
Transcription, mRNA processing, translation, or protein stability post translation
What are the possible genotypes of β-Thalassaemia Minor
β/β⁰
β/β⁺
β0 = absent
β+ = reduced
What are the symptoms of β-Thalassaemia Minor
Often asymptomatic or mild microcytic anaemia.
β-Thalassaemia Major: What are the possible genotypes
β⁰/β⁰
β⁺/β⁺
β⁰/β⁺
Both copies of Chr 11 affected
What is the clinical outcome of β-thalassaemia Major
Severe anaemia requiring lifelong transfusions.
What genes encode α-globin and where are they located?
HBα1 and HBα2, both on chromosome 16 (four copies total - 2 on each chromosome).
What are the four clinical categories of α-thalassaemia?
α⁺: α-thalassaemia minima / silent carrier (one defective locus)
α0: α-thalassaemia minor/trait: (2 defective loci). Often clinically asymptomatic / mild cryptic symptoms (mild hypochromic microcytosis, mild anaemia.
HbH disease: (3 defective loci) Beta chain excess and production of HbH (4 x B-globins)
α-thalassaemia major aka HbBart: (4 defective loci)[Hydrops faetalis] No oxygen released to tissues and foetus dies – most serious form
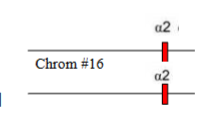
Which α-thalassaemia is this
α0-thalassamia ( 2 defective loci)
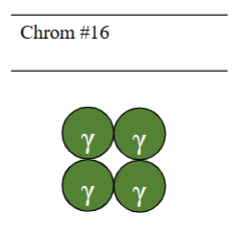
Which α-thalassaemia is this
α-thalassaemia major
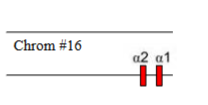
Which α-thalassaemia is this
α-thalassaemia trait
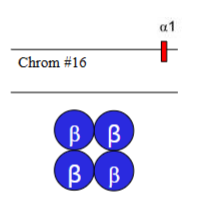
Which α-thalassaemia is this
HbH disease
What races are more at risk of α-thalassaemia
HbH disease (3 defective loci ) is most common in Asian heritage

Hb genes on Chr 11 and Chr 16 are located in a 5’ to 3’ direction what order
the order in which they are expressed during embryonic/fetal life
The LCR controls the expression of beta globin genes in cis. It is located where in relation to the globin genes on Chr 11
It is located 1000s of base pairs upstream (5’) of the globin genes on Chr 11