Microbio Module 3
1/40
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
41 Terms
Micrometer and Nanometer measurements
micrometer: 10^-6m
nanometer: 10^-9m
how tiny can human eyes vs light microscopes vs electron microscopes see
human: >200 µm
light: >0.2 µm
electron: <1nm
Resolution
distinguish 2 things as separate (how close can 2 objects be before they look like 1?)
higher resolution = more detail
Contrast
difference in light between object and background
higher contrast = easier to see
Brightfield Microscope
most common
light passes THROUGH specimen
light source —> condenser —> speciment —> objective lens —> ocular lens/eyepiece
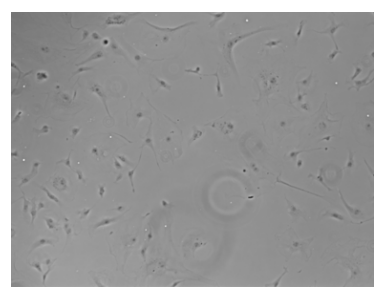
disadvantages of Brightfield
unstained cells = basically invisible
requires staining
staining kills/fixes cells
magnification formula
objective x ocular
illuminator
light source
Iris diaphragm
controls amount of light entering condenser
condenser
focuses light through specimen
objective lens
primary lens that magnifies specimen
closest to specimen
provides most magnification
body
transmits image from objective to ocular lens using prisms
ocular lens (eyepiece)
remagnifies image formed by objective lens
usually 10x
Phase Contrast Microscope
Enhances tiny differences b/w cell and its background
best for LIVE cells (no staining needed)
can look at motility + internal structures
more specialized than brightfield
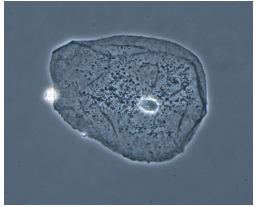
Dark Field Microscope
light reflects OFF speciment at an angle
(GREAT contrast) black background, bright/glowing specimen
best for THIN organisms
can’t see internal structures
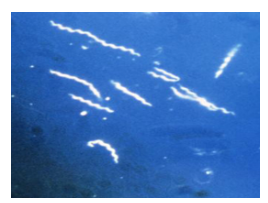
Fluorescence Microscope
uses fluorophores (flourescent dyes/proteins)
uses UV light —> excites fluorophores at diff wavelengths —> glow diff colors (wide variety)
dark background, bright colored objects
ex: GFP, RFP, YFP
used to locate specific proteins, detect antibodies
can’t visualize intracellular structures
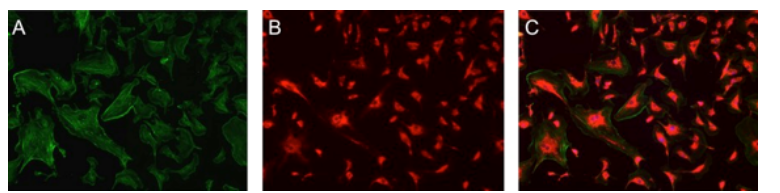
how are fluorescent proteins expressed in a cell alone
nonspecifically illuminating cell as whole
linked (coupled) to normal protein of interest
tags on molecules/antibodies —> designate presence/absence of specific protein target
fluorescence detected: protein present
no fluorescence: protein absent
Confocal (Laser Scanning) Microscope
fluorescence microscope + 3D imaging
high-resolution, 3D, multiple cell layers, light = laser light
layer scans 1 thin layer at a time —> computer stacks images together —> 3D image
GREAT resolution + contrast
3D
individual organelles visible
Why use Electron Microscopes
light microscopes stop at 0.2 µm
viruses < 0.2 µm
electron microscopes can see viruses (resolution <1nm) = 200x better than light microscopes
uses beams of e-
e- wavelength < light wavelength
shorter e- wavelength —> higher resolution
Disadvantages of electron microscopes
expensive, complex, labor intensive
sample must be DEAD
process may alter cell structure
TEM
electron passes THROUGH thin slices of specimen (transmission = through)
coated in preservatives, heavily treated
sample in b/w e- beam source and detector
2D
subcellular structures (organelles), viruses
SEM
electrons bounce OFF the surface (scanning = surface)
specimen coated w/ gold or palladium (e- coated)
3D appearance
surface details, external shape
STEHM
HIGHEST resolution (35 pm)
protein surfaces
subatomic structures
stains used to examine
b/c most cells = transparent
chemical dye = makes microorgs easier to see
tissues
specific cell types
organelles w/in a cell
differential stain
staining technique that separates specimens into subgroups
usually uses at least 2 dyes
gram
acid-fast
giemsa
simple stain
uses 1 positive charged dye
examine: size, shape, arrangement
gram stain
developed by Hans Christian Gram (1884)
distinguishes thickness of peptidoglycan cell wall (thick/thin)
Gram Positive
very thick peptidoglycan cell wall (overlapping layers)
permeable
no outer membrane
+crystal violet —> +iodine —> stable complex —> thick peptidoglycan traps dye
—> cell stays PURPLE
Gram negative
thin peptidoglycan cell wall
outer membrane = made of LPS (lipopolysaccharides)
+crystal violet —> +iodine —> thin wall can’t trap dye —> alcohol wash removes dye —> LPS loses color —> +safranin —> cell becomes PINK
Gram Stain Steps
Color It And Save (CIAS)
crystal violet — primary stain
iodine — forms stable complex
alcohol — decolorization wash
safranin — counterstain
why does alcohol matter
removes crystal violet from gram negative
BUT NOT
gram positive (b/c it has thick peptidoglycan)
heat fixation
cells must stick to slide before staining —> prevents cells from washing away BUT kills microorg (can’t observe motility)
air dry sample
pass through flame (cell attaches)
can stain
Chemical fixation
instead of heat, chemicals can attach cells (still kills cells)
ex:
formaldehyde
ethanol
methanol
wet mount
opposite of Gram stain
wet = alive: no fixing, no heating, no killing
can observe movement, behavior, motility
drop of liquid on glass slide
cover slip
Simple stain
1 positively charged dye to stain organism
quickly determine: shape, size, arrangement
ex:
methylene blue
crystal violet
safranin
fuchsin
why positive dye?
bacterial membrane = neg charge
opposites attract, positive dye sticks
Negative stain
opposite of simple stain
stain everything BUT organism
dye = nigrosin (India Ink) = neg charge
dye + membrane = both neg charge, like repels, dye can’t attach
positive vs negative dye
negative = background gets color
positive = cell gets color
acid-fast stain
some bacteria do not Gram stain well
esp Mycobacterium TB b/c very thick, lipid-rich protective membrane
primary stain = Carbolfuchsin (red)
alochol wash (acid-fast stay red)
counter stain = methylene blue
non-acid fast-become blue
TB (acid-fast)= red
healthy background = blue
Giemsa Stain
combined with Wright’s stain
used for blood smears
Malaria
blood parasites
human blood cells: purple
bacteria: pink
structural stains
features of bacterial cells
endospore
capsule
flagella