Genetics: Exam 3
1/28
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
29 Terms
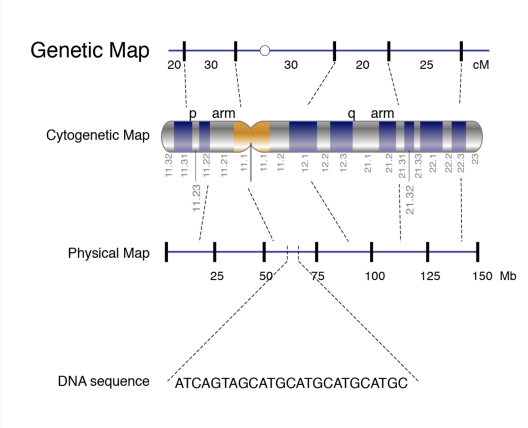
Genetic (linkage) maps vs. physical maps
Genetic (linkage) maps- approximate location of genes based on recombinant rates
Physical maps- more accurate; higher resolution than genetic maps
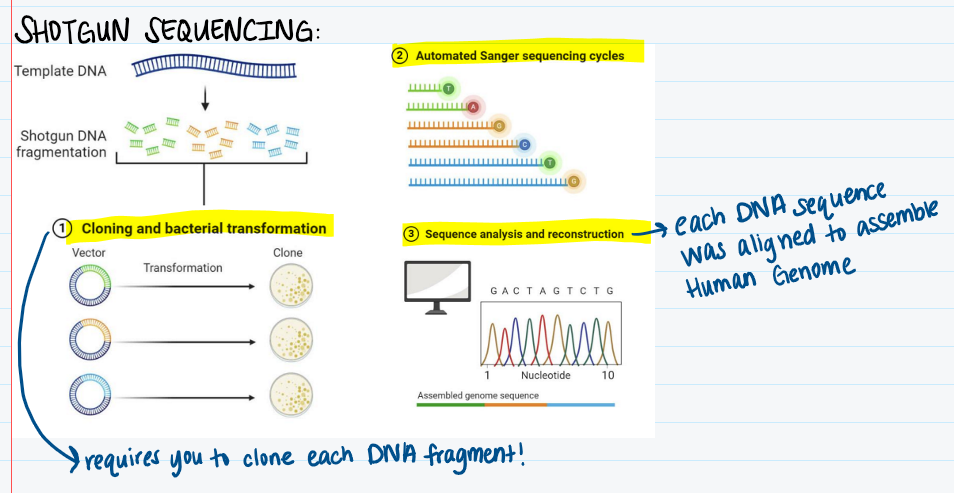
Describe shotgun sequencing.
Breaks up DNA into smaller fragments that are reassembled to reconstruct the original sequence
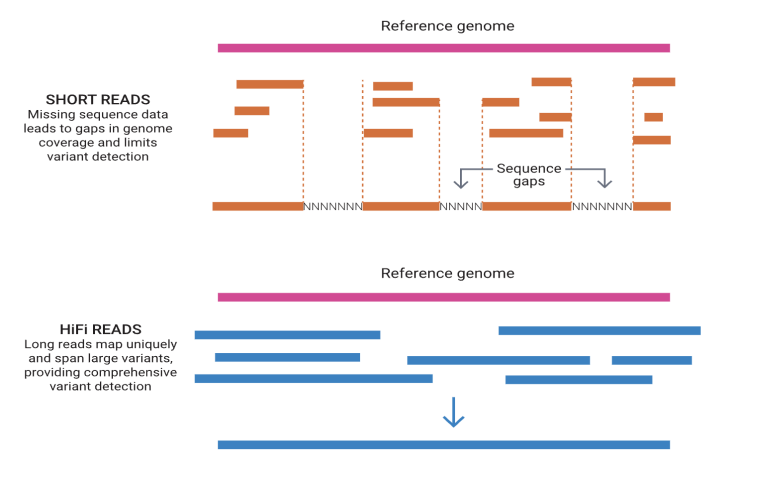
Short vs. long reads
Short reads- tend to have gaps in genome
Long reads- spans long variants; more comprehensive
2nd next-generation DNA sequencing
a short read
increased efficiency
a nucleotide is bound to a different fluorescent color which helps you to read the DNA sequence
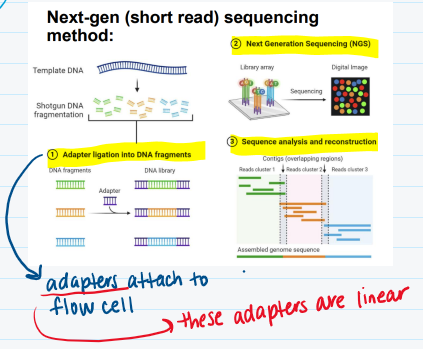
3rd generation next-gen sequencing
long read sequence
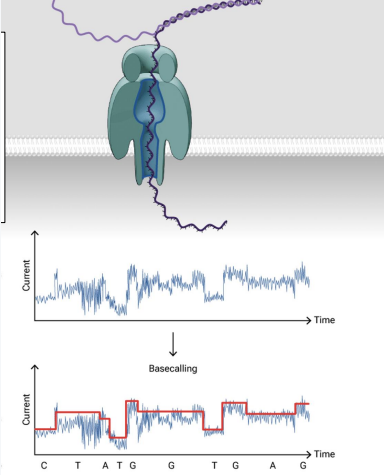
There’s a nanopore method and PacBio Hifi method
Nanopore method
has the longest reads
reads nucleotides based on ELECTRICAL charge
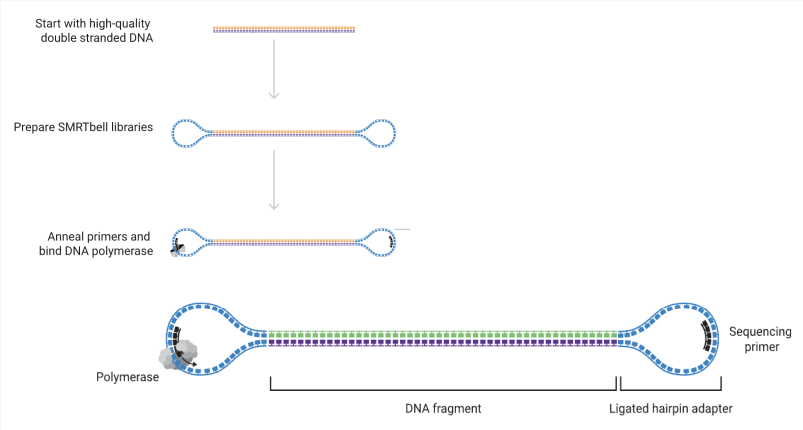
PacBio Hifi method
relies on sequencing a circular DNA molecule
adds circular adapter to ends of DNA sequence
HiFi is more accurate than the nanopore method
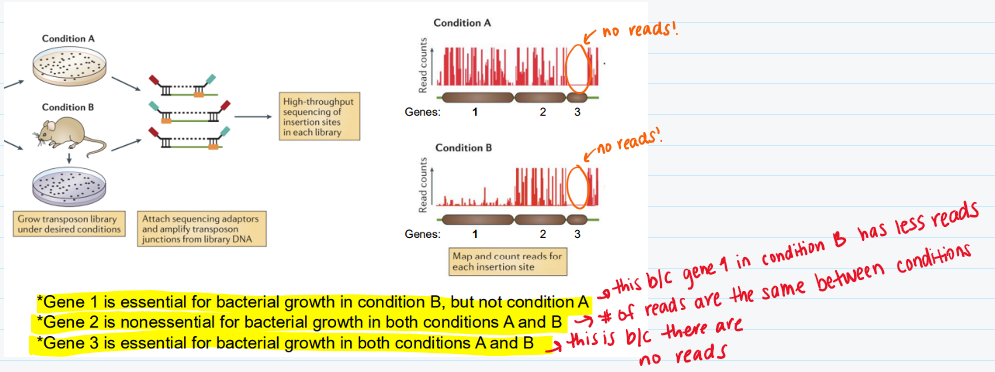
List the key steps for a Tn-Seq
1) Select your transposon type
Ex: bacteria, Drosophila, mice
2) Select your selectable marker
Ex: antibiotic resistance gene, red eye color, and GFP
3) Clone transposon & selectable marker. Don’t forget the transposase!
4) Screen for transposition in your organism.
5) Expose transposed organisms to different conditions
6) Isolate genomic DNA from organisms grown in different conditions
7) Fragment genomic DNA
8) Ligate (combine) DNA fragments with a transposon specific adaptor
Ex: transposon is designed to have restriction enzymes site (ex:Msel) allowing DNA to be digested and ligated to sequencing adaptor
9) Proceed with genome-wide DNA sequencing method

How to determine where the transposon inserted?
To find where the transposon inserted, you want to do a short read sequencing
You could also use a long read, if there are repetitive nucleotide sequences
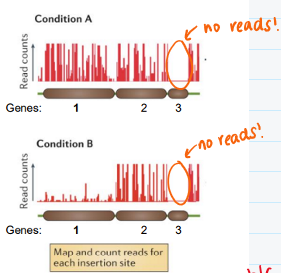
How can you interpret the function of genes 1, 2, and 3 based on the number of reads?
(look at the image)
Sanger vs. next-gen sequencing
Sanger sequencing- only does 16 reactions per gel; lacks 3’ OH; adds fluorescently labeled ddNTPs
Sanger sequencing is preferred, if you ONLY want to know the sequence of one or a few genes, instead of the whole genome!
Next-gen sequencing- 10,000 reactions per slide
What is genome wide association study (GWAS)?
An approach that rapidly scans for markers, like SNPs, across the genome
Once an marker (SNP) is identified, you can determine where it’s located and the DNA sequence associated with the trait
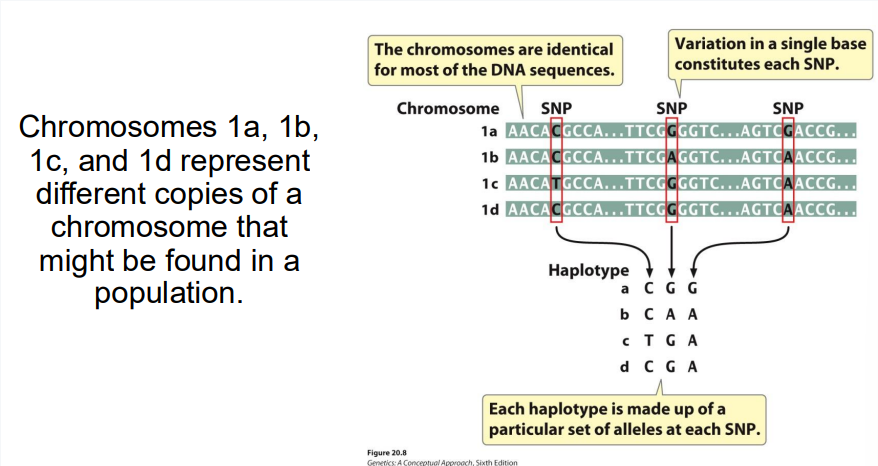
What is an SNP?
Site on genome where individual members of a species differ in a single base pair
Haplotype
Specific set of SNPs and other genetic variants observed on a single chromosome or part of chromosome
What if an SNP maps to a human gene of unknown function?
1) First, determine where the SNP is located in the gene.
This can include a protein coding region (ORF), regulatory region (promoter/enhancer), intron (UTRs)
2) Search for similar genes in other species that have known function
Describe homologous genes, orthologs, & paralogs.
Homologous- genes that are evolutionarily related
Orthologs- homologous genes in different species
Paralogs- homologous genes arising by duplication in SAME organism
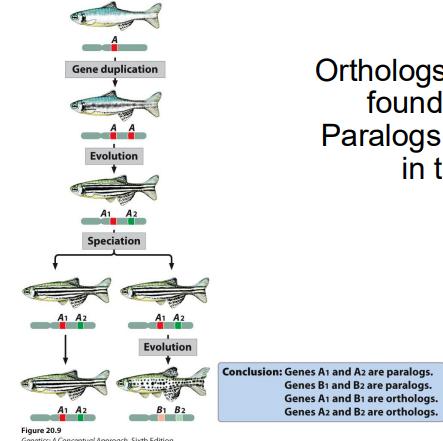
What are some mechanisms of genome evolution?
1) Mutation- causes the majority of genome evolution
2) Gene duplication- another source of genome evolution & gives rise to paralogs
3) Gene deletion
4) Exon shuffling- shuffling of genes through recombination
Synonymous mutations vs. nonsynonymous mutations
Synonymous mutations- do NOT alter amino acid sequence
Ex: a silent mutation
Nonsynonymous mutations- ALTER amino acid sequence
Neutral-mutation hypothesis
Individuals with different molecular variants have EQUAL fitness
Balance hypothesis. Give an example
Genetic variations in populations that FAVOR variation
Ex: overdominance- where the heterozygote has higher fitness than the homozygote
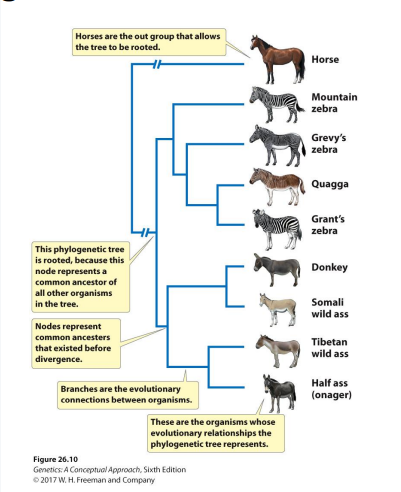
Define phylogeny and phylogenetic tree.
Phylogeny- evolutionary relationship among a group of organisms
Phylogenetic tree- a graphical representation of the evolutionary relationships among a group of organisms
Define transcriptomics. List single transcript and whole transcriptome methods.
Transcriptomics- measures all RNA molecules transcribed in genome
Single transcript: In-site hybridization (in-situ), northern blot, reverse transcription polymerase reaction (RT-PCR)
Whole transcriptome: cDNA microarray & RNA-seq
In-situ hybridization (ISH)
Allow for precise localization of a specific segment of nucleic acids in a cell/tissue section
RNA sequences are based on binding of a probe
Probes are labeled with markers (ex: radioactive isotopes, fluorescence, etc.)
Advantage: helps resolve which cell type expression is present

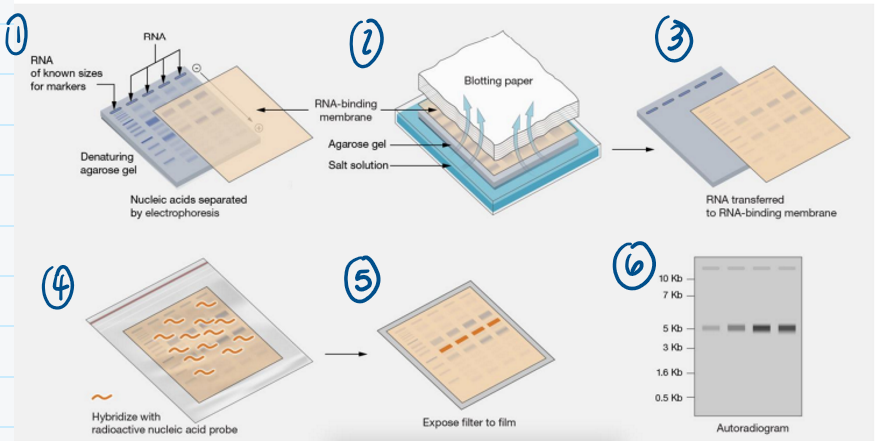
Northern blot
Begins with isolating RNA from cell, tissue, etc.
Then, RNA gets ran on an agarose gel
Uses electrical current to transfer RNA onto RNA-binding membrane
Probe for whatever gene of interest
If mRNA is present, probe will bind and you expose filter to film
The more intense the band is, the more the gene is expressed
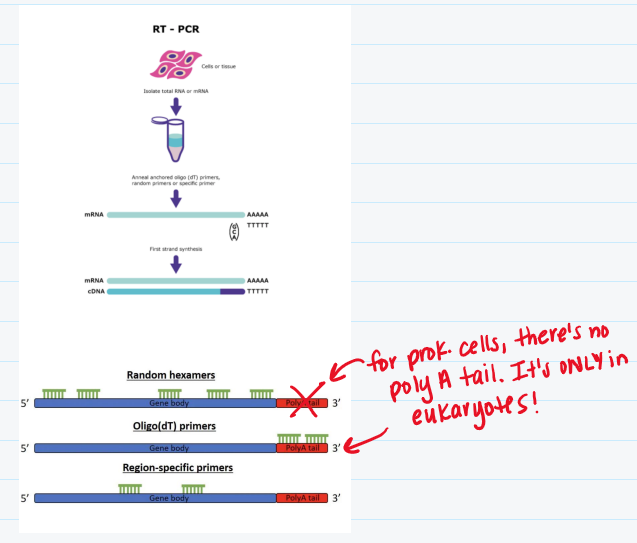
Reverse transcription polymerase chain reaction (RT-PCR)
Isolate RNA from cell/tissue/organism
Reverse transcribe mRNA to complementary DNA (cDNA)
To do this, you need reverse transcriptase, dNTPs, & DNA primer
For eukaryotic cells, you need Oligo T (primer)
For prokaryotic cells, you need random hexamers (primer)
Amplify gene of interest using PCR with gene specific primers, DNA polymerase & dNTPs
Take amplified sequence and run it through gel electrophoresis to resolve RT-PCR DNA sequence
Intensity of band = expression of transcript (like in Northern blot)
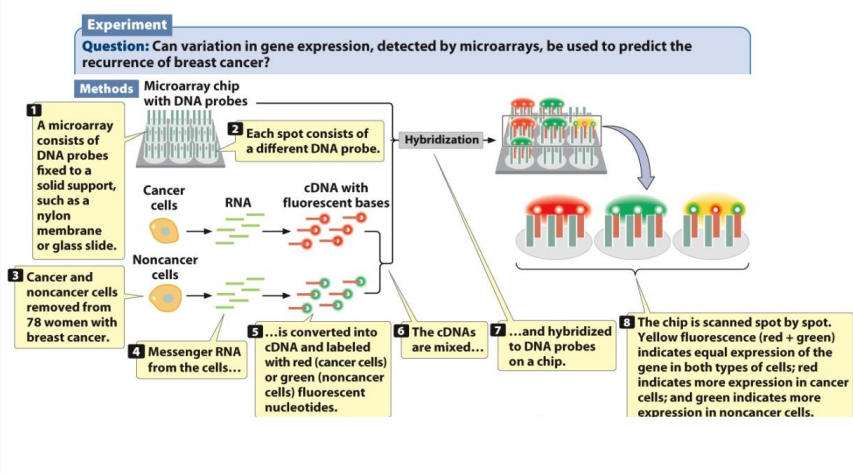
DNA Microarray (also list its disadvantages)
It’s a chip that allows thousands of transcripts to be determined simultaneously
1) You need cells from 2 or more different conditions to isolate total RNA
2) Reverse transcribe mRNA to cDNA
3) Label different conditioned cells with fluorescent markers
4) cDNAs are mixed and hybridized to DNA probes on a chip
5) Then, the chip is scanned
If there are equal amounts of expression in BOTH cell types of different conditions (yellow fluorescence)
More expression of one cell types (ex: cancer cell)→ red fluorescence
More expression of another cell type (ex: noncancer cell)→ green fluorescence
Disadvantages: low sensitivity, low dynamic range, no alternative splicing
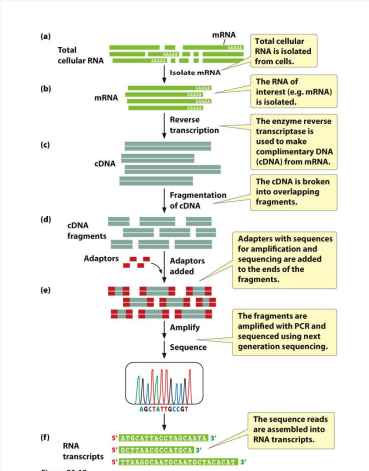
RNA Sequencing (RNA-seq)
Allows you to identify ALL mRNA transcripts present in cell
1) Isolate total RNA from cells (specific whether prokaryotic or eukaryotic)
2) Select for type of RNA (in this case, mRNA)
3) Reverse transcribe RNA into cDNA using DNA primers, dNTPs, and reverse transcriptase
Remember, for eukaryotic cells → Oligo T primer
For prokaryotic cells→ Random hexamer primer
4) cDNA gets fragmented and adaptors are added
5) Lastly, sequence using short read sequencing for bacteria and long read sequencing for eukaryotes
Advantages: high sensitivity & high dynamic range
Why do you use short read sequencing for bacteria and long read sequencing for eukaryotes?
This is because long read sequencing allows alternatively spliced transcripts to be identified. Bacterial transcripts are not spliced.
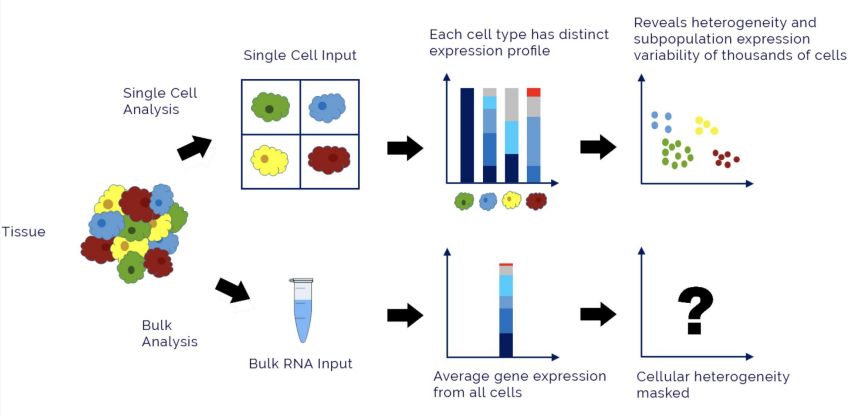
Single cell vs. bulk RNA-seq
Involves the same steps as in RNA-seq; however….
Single cell separates each INDIVIDUAL cell, while bulk keeps cells in a GROUP