CM2-CM5: Bases génétiques des maladies héréditaires
1/47
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
48 Terms
1 Definition
Qu’est-ce qu’une maladie mendélienne ?
Quelle est la différence entre autosomes et gonosomes ?
Qu’est-ce qu’un locus et des loci homologues ?
Quelle est la différence entre gènes homologues et allèles ?
Combien d’allèles peut-on retrouver à un locus donné en population générale et chez un individu ?
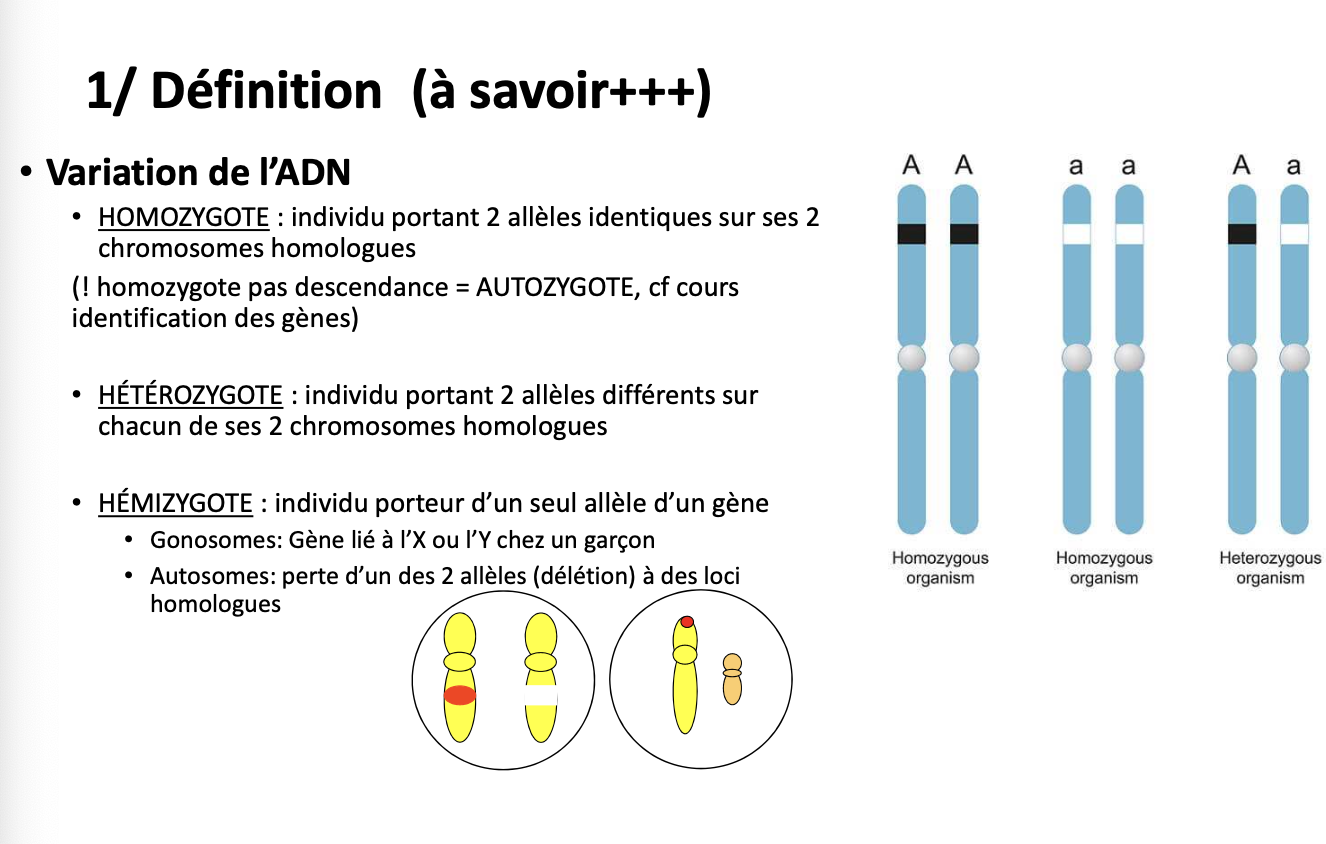
Quelle est la différence entre homozygote, hétérozygote et hémizygote ?


Maladie mendélienne : c’est une maladie monogénique dont la transmission suit les lois de Mendel.
Autosomes : 22 paires de chromosomes identiques dans les deux sexes.
Gonosomes : chromosomes sexuels, X et Y.Locus : emplacement d’un segment d’ADN (par exemple un gène) sur un chromosome.
Loci homologues : même position sur les deux chromosomes d’une paire homologue.Gènes homologues : gènes occupant le même locus sur une paire de chromosomes homologues.
Allèles : versions alternatives d’un même gène ou marqueur, s’excluant mutuellement au moment de la méiose.Population générale : il peut exister de nombreux allèles différents à un locus donné.
Chez un individu :Autosomes → 2 allèles par locus.
Gonosomes → 2 allèles si fille (XX), 1 seul si garçon (XY).
Homozygote : 2 allèles identiques sur une paire de chromosomes homologues.
Hétérozygote : 2 allèles différents sur une paire de chromosomes homologues.
Hémizygote : un seul allèle pour un gène (ex. gène lié à X chez un garçon, ou perte d’un allèle par délétion).
1 Definition
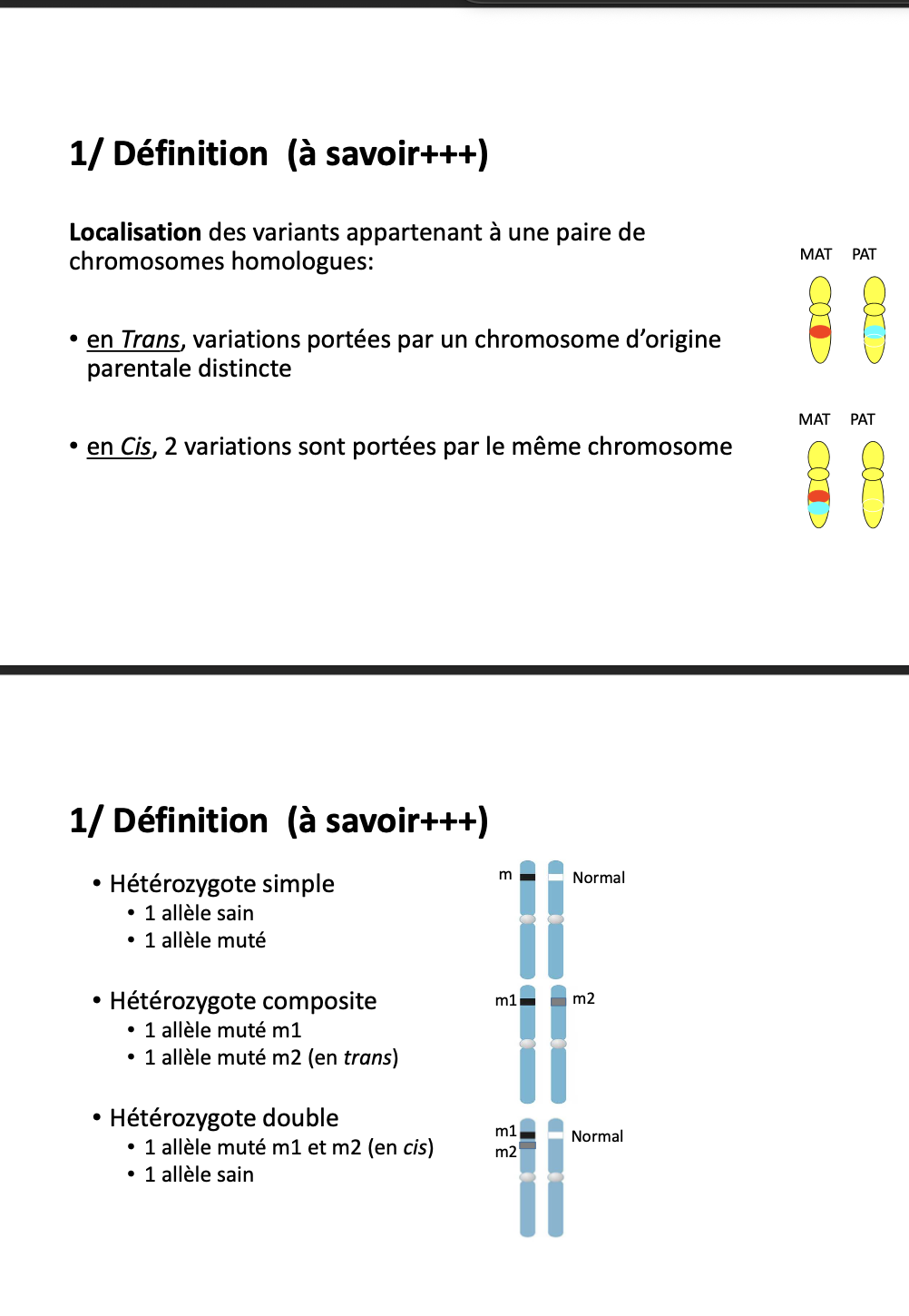
Quelle est la différence entre une localisation en trans et en cis des variants génétiques ?
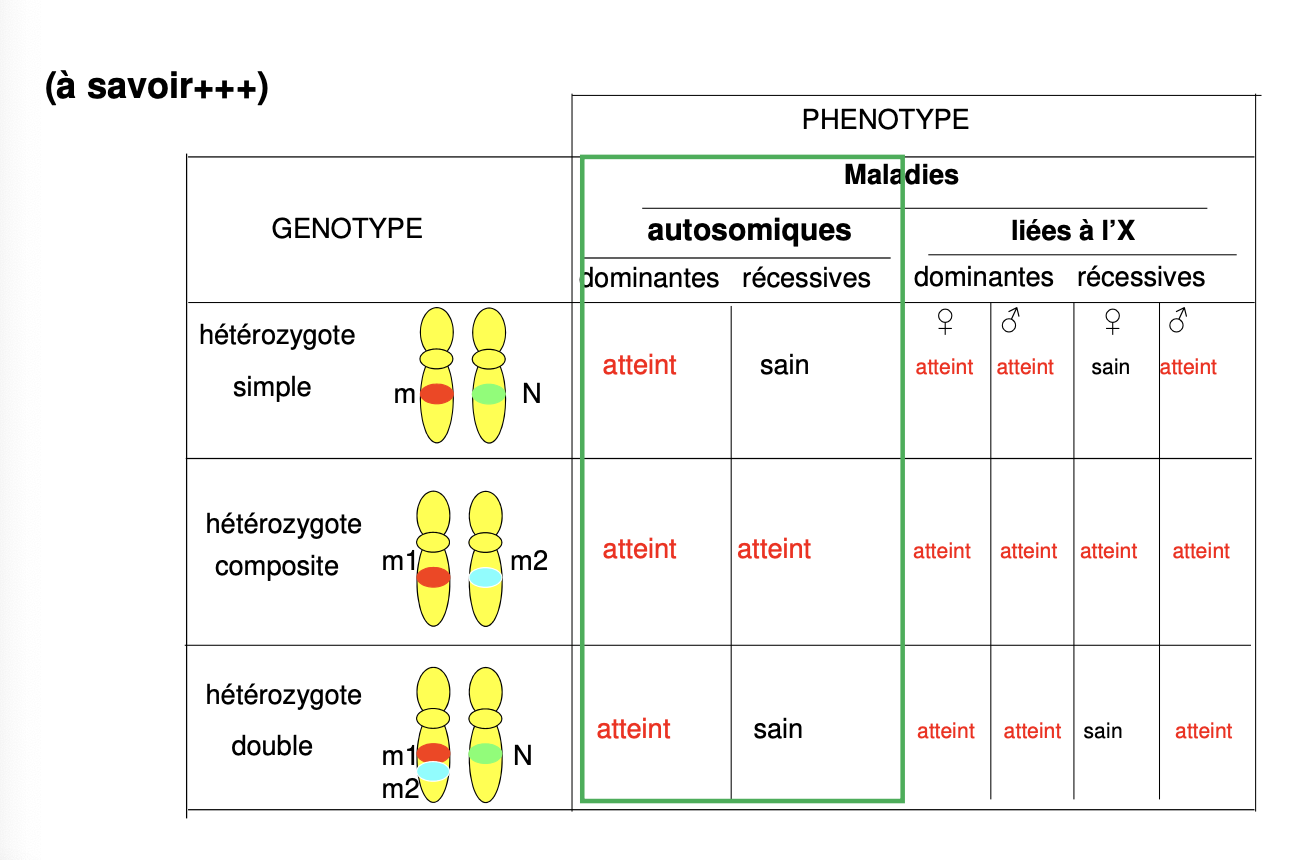
Qu’est-ce qu’un hétérozygote simple ?
Qu’est-ce qu’un hétérozygote composite ?
Qu’est-ce qu’un hétérozygote double ?
Localisation des variants :
En trans : chaque variant est porté par un chromosome d’origine parentale différente (ex. mutation maternelle sur un chromosome, mutation paternelle sur l’autre).
En cis : deux variants sont situés sur le même chromosome (provenant du même parent).
Hétérozygote simple : individu qui possède un allèle sain et un allèle muté.
Hétérozygote composite : individu qui possède deux allèles mutés différents (m1 et m2) localisés sur des chromosomes homologues distincts (configuration en trans).
Hétérozygote double : individu qui possède un chromosome avec deux mutations (m1 et m2 en cis*) et un autre allèle sain.
1 Definition
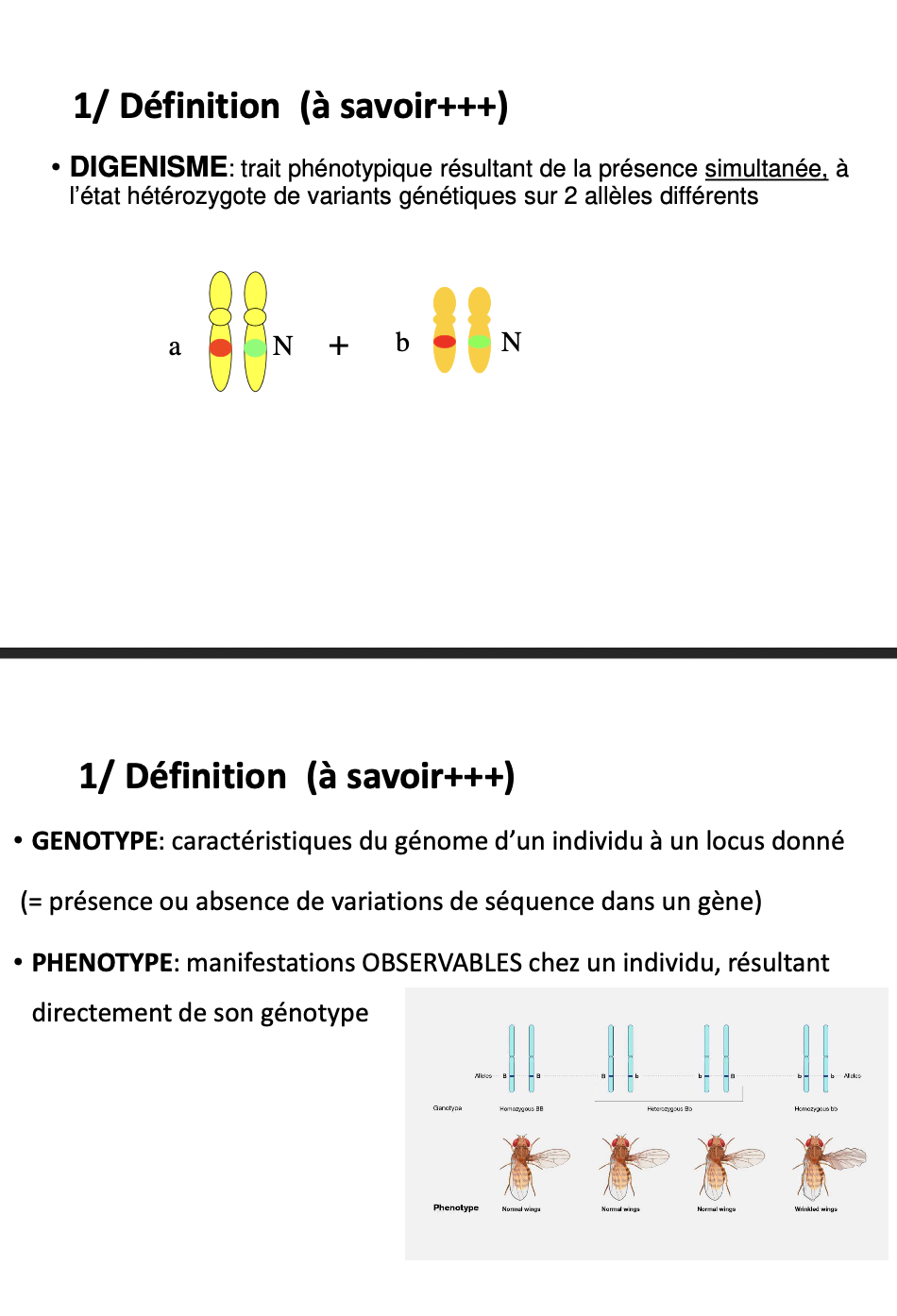
Qu’est-ce que le diginisme ?
Quelle est la différence entre génotype et phénotype ?

Diginisme :
C’est un trait phénotypique qui résulte de la présence simultanée de deux variants génétiques différents portés en hétérozygotie sur deux gènes distincts (ou deux allèles différents).
Exemple : un individu présente une mutation a sur un gène d’un chromosome, et une mutation b sur un autre gène → leur combinaison produit un phénotype pathologique.
Génotype vs Phénotype :
Génotype : ensemble des caractéristiques génétiques d’un individu à un locus donné (présence ou absence de variations de séquence).
Phénotype : ensemble des caractères observables résultant directement de l’expression du génotype (morphologie, symptômes, traits cliniques…).
Qu’est-ce qu’un gène pléiotrope ?
Quels sont les mécanismes expliquant la pléiotropie ?
Donne un exemple clinique de gène pléiotrope.
Définition d’un gène pléiotrope :
C’est un gène dont les mutations entraînent des modifications phénotypiques multiples, touchant des caractères sans relation apparente.
Exemple : une seule mutation peut provoquer des anomalies du squelette, des yeux et du système cardiovasculaire.
Mécanismes :
Mutation dans une protéine ubiquitaire (exprimée dans plusieurs tissus).
Mutation dans un gène d’expression précoce durant le développement embryonnaire → impact sur plusieurs organes en formation.
Exemple clinique :
Syndrome de Marfan (mutation du gène FBN1) → entraîne : grande taille, luxation du cristallin, fragilité de l’aorte (dissection).
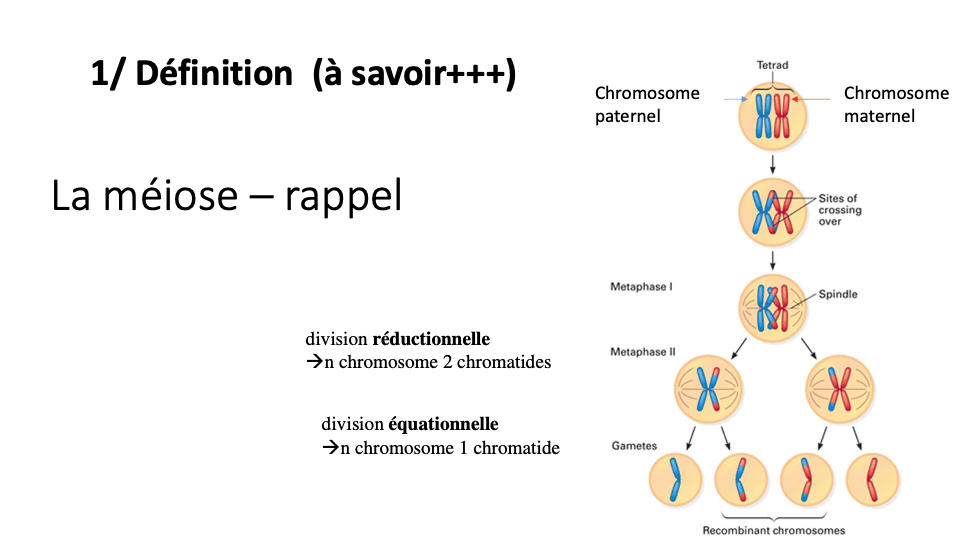
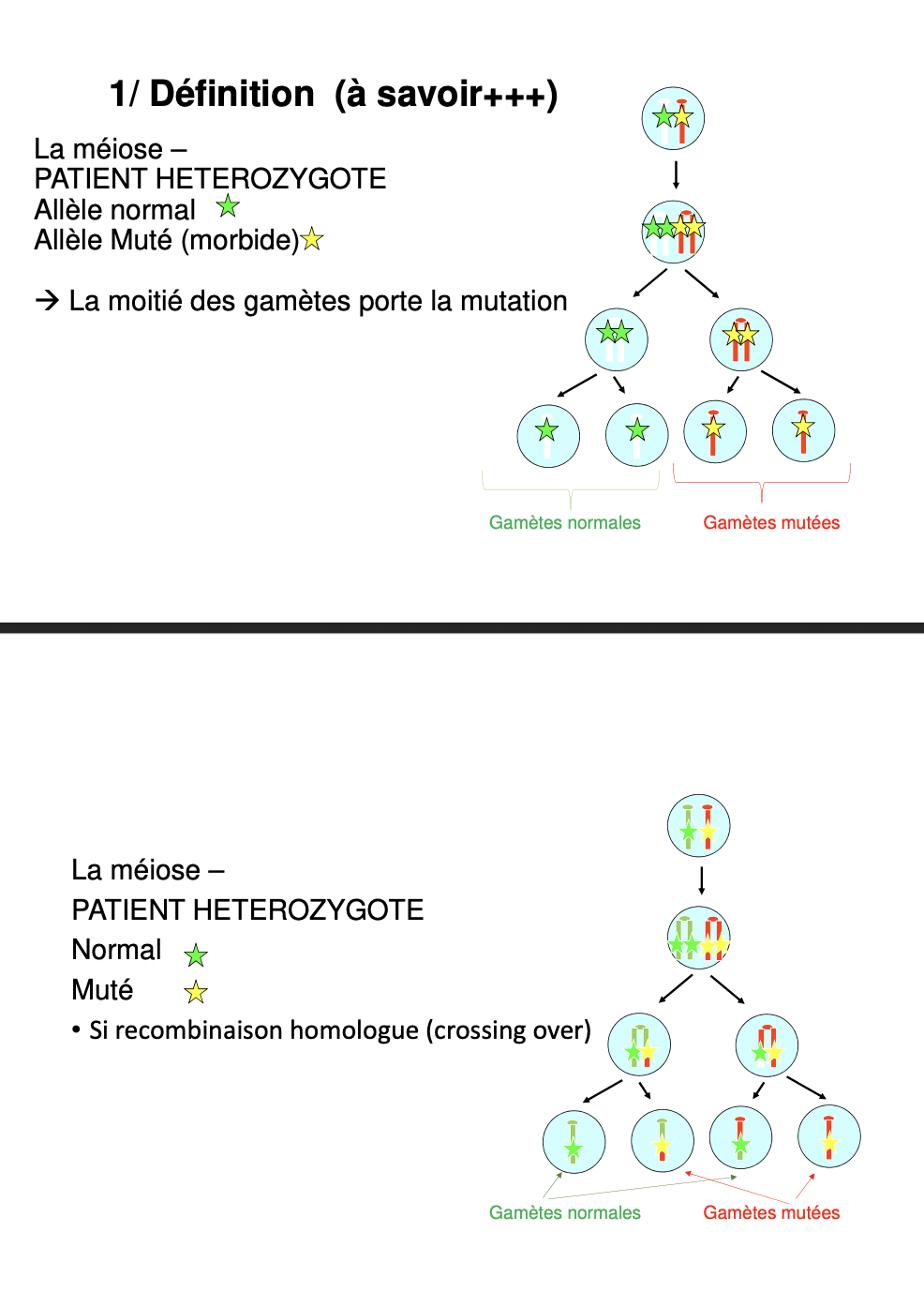
Quelles sont les deux grandes étapes de la méiose et leurs caractéristiques principales ?
Quelle est la différence entre une division réductionnelle et une division équationnelle ?
Quel est le rôle du crossing-over pendant la méiose ?
Les deux grandes étapes de la méiose :
Méiose I (division réductionnelle) : séparation des chromosomes homologues → produit deux cellules avec n chromosomes, chacun à 2 chromatides.
Méiose II (division équationnelle) : séparation des chromatides sœurs → produit quatre cellules haploïdes (n chromosomes, chacun à 1 chromatide).
Différence entre réductionnelle et équationnelle :
Division réductionnelle (Méiose I) : réduit le nombre de chromosomes (2n → n) tout en gardant 2 chromatides par chromosome.
Division équationnelle (Méiose II) : conserve le nombre de chromosomes (n → n) mais sépare les chromatides (n chromosomes à 1 chromatide).
Rôle du crossing-over :
Se produit en prophase I entre chromatides homologues.
Permet un échange de matériel génétique → diversité génétique des gamètes.
Contribue à la recombinaison et à l’unicité de chaque individu.
2 Heredite mendelienne
-automisomique (dominante)

quand parle on de mosaique germinale? comment l’avoir?

Hérédité mendélienne – Autosomique récessive (AR) + pseudodominance & Hardy–Weinberg
Partie 1 — Questions (seules)
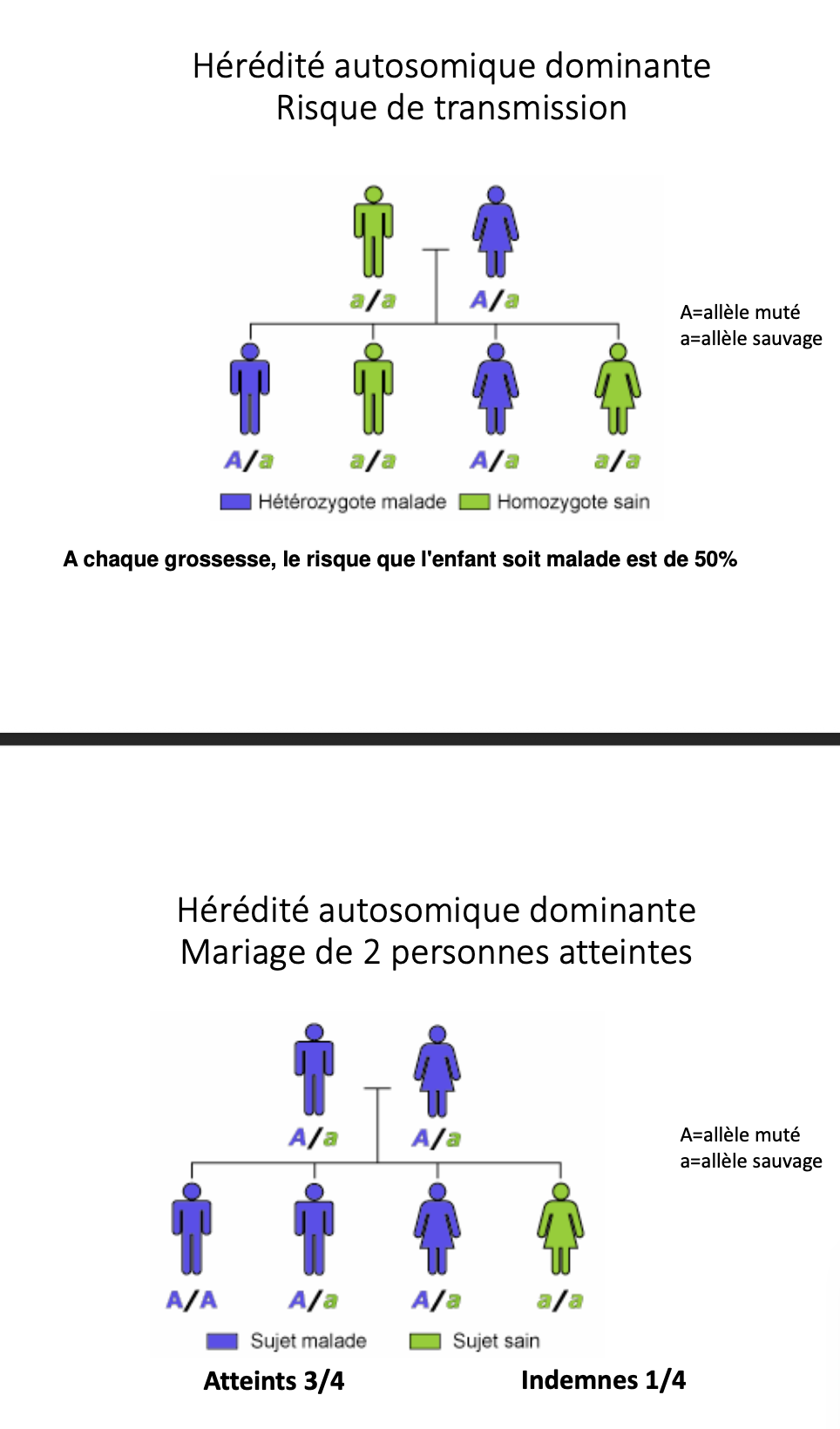
Que veut dire autosomal récessif ?
Quelle est la répartition homme/femme des sujets atteints ?
Quel est l’aspect typique d’un pedigree AR (profil de transmission) ?
Quels génotypes et risques à chaque grossesse pour Aa × Aa ?
Quel risque d’enfant atteint pour aa × AA ?
Quel risque d’enfant atteint pour aa × Aa ?
Quel risque d’enfant atteint pour aa × aa ?
Gène sur un autosome ; la maladie s’exprime quand on porte deux allèles mutés (aa) (ou hémizygote par délétion).
♀ = ♂ (atteinte égale).
Transmission horizontale : sujets atteints souvent dans une seule génération ; parents sains porteurs ; consanguinité fréquente.
Aa × Aa : 25 % aa (atteint), 50 % Aa (porteur sain), 25 % AA (sain non porteur) — à chaque grossesse.
aa × AA : 0 % atteints, 100 % porteurs (Aa).
aa × Aa : 50 % atteints (aa), 50 % porteurs (Aa).
aa × aa : 100 % atteints.
Hérédité mendélienne – Autosomique récessive (AR) + pseudodominance & Hardy–Weinberg
Comment dépiste-t-on les hétérozygotes porteurs ?
Rappeler la loi de Hardy–Weinberg et l’estimation de la fréquence des porteurs pour une maladie AR rare.
Qu’est-ce que la pseudodominance ?
Quels indices orientent vers l’AR dans un arbre généalogique ?
Citer des exemples de maladies AR fréquentes.
Message clé de conseil génétique à des parents d’un enfant AR atteint.
En AR, un hétérozygote (Aa) est-il malade ?
Pourquoi la consanguinité augmente-t-elle le risque de maladie AR ?
Analyse moléculaire (génétique) ; parfois phénotype mesurable (ex. activité enzymatique ≈ 50 %).
p+q=1; atteints = q^2 ; porteurs = 2pq≈2q si q≪1. Ex : si q^2 ≈1/4500 ⇒ q≈1/67⇒ porteurs ≈ 1/33.
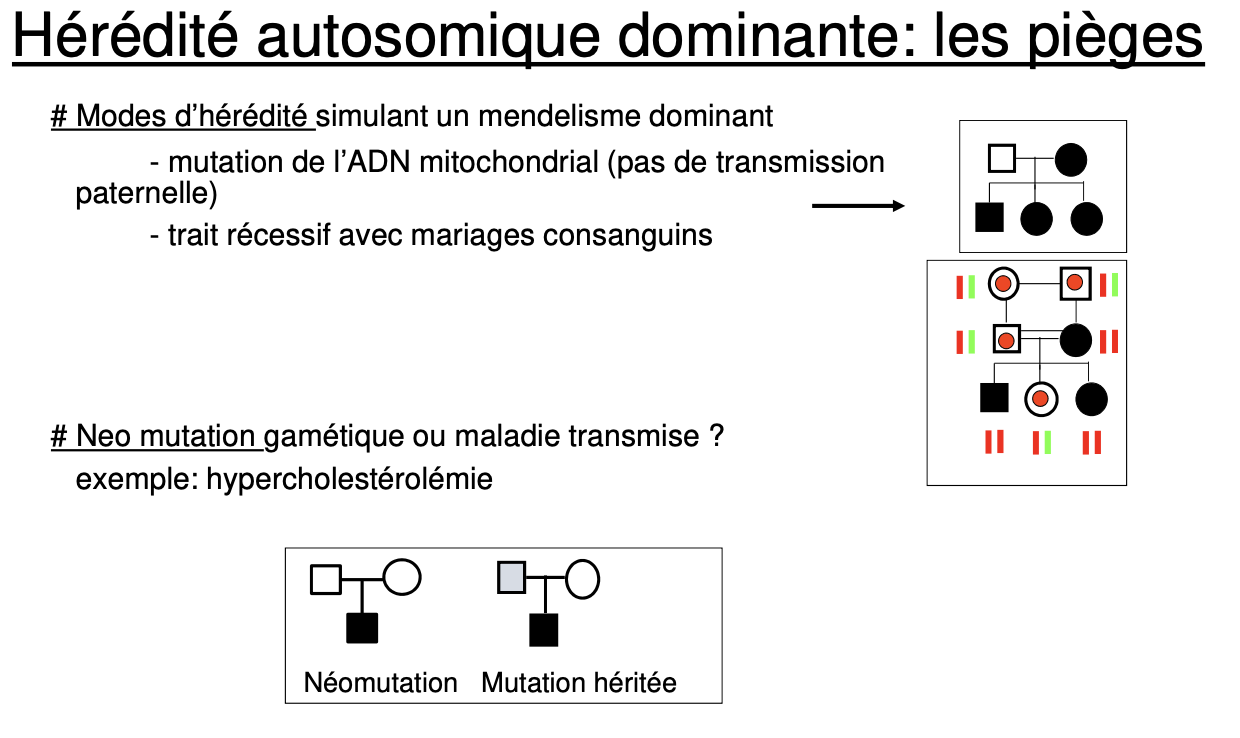
Apparence “dominante” (atteints sur plusieurs générations) due à forte fréquence de porteurs/consanguinité ⇒ plus d’unions Aa × Aa.
Parents non atteints d’un enfant atteint, fratries touchées, consanguinité, sexes atteints également.
Mucoviscidose, drépanocytose, phénylcétonurie, SMA, Tay-Sachs, etc.
Les deux parents sont porteurs obligatoires (Aa) (sauf exception) ; risque de récurrence 25 % à chaque grossesse.
Non (en général) : un hétérozygote Aa est porteur sain.
Elle augmente la probabilité que les conjoints portent la même mutation, donc plus de couples Aa × Aa.
1) Perte de fonction
- haploinsuffisance
- un cas particulier: effet dominant négatif
les deux macanismes
quest ce que l’haploinsufissnace(exemple avec prot trimerique)? l’effet dominant négatif?


Hérédité autosomique dominante — Bases moléculaires : Perte de fonction
(sous-mécanismes : haplo-insuffisance & effet dominant négatif)
Partie questions
Que signifie « perte de fonction » d’un gène/protéine ?
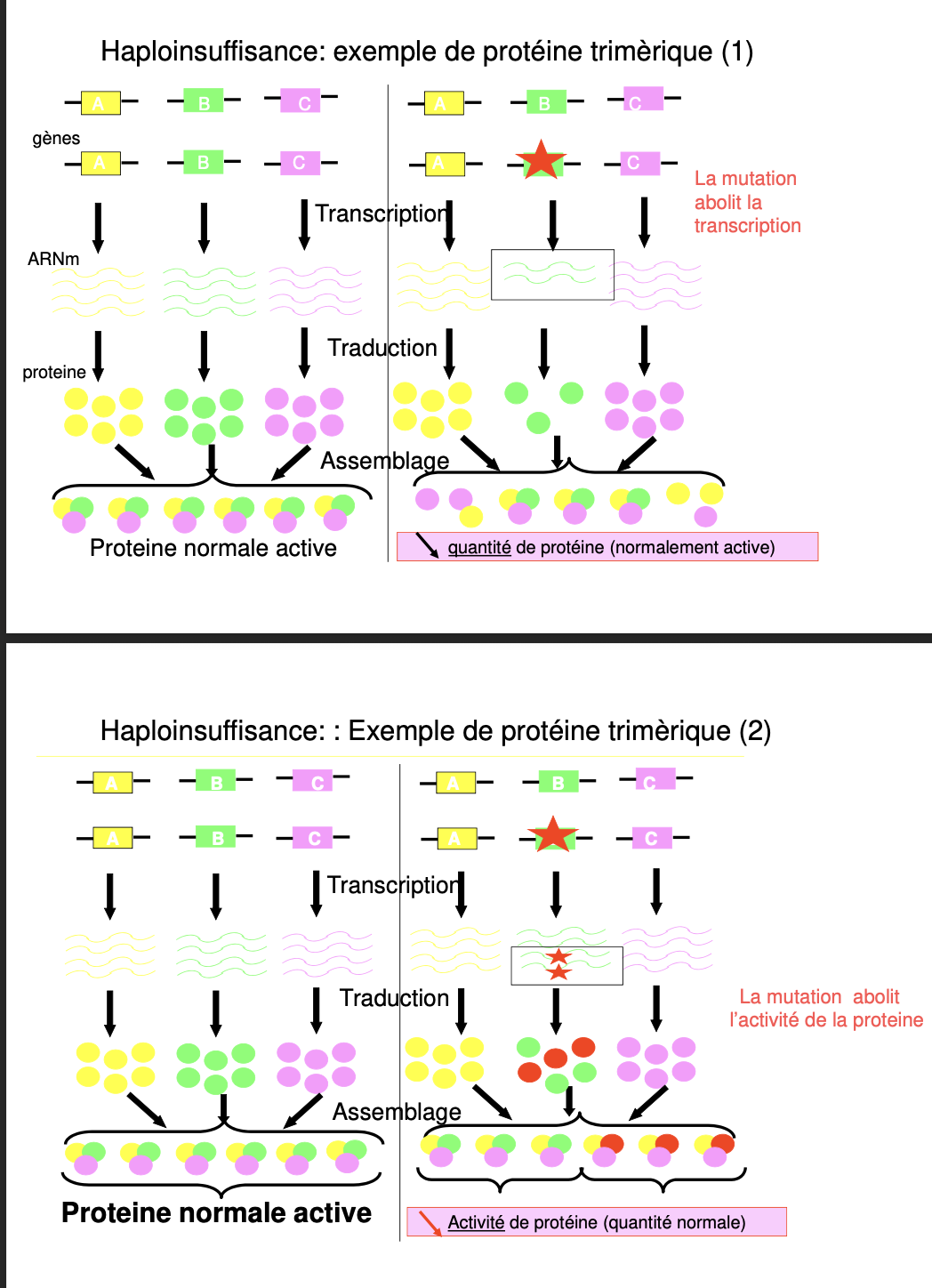
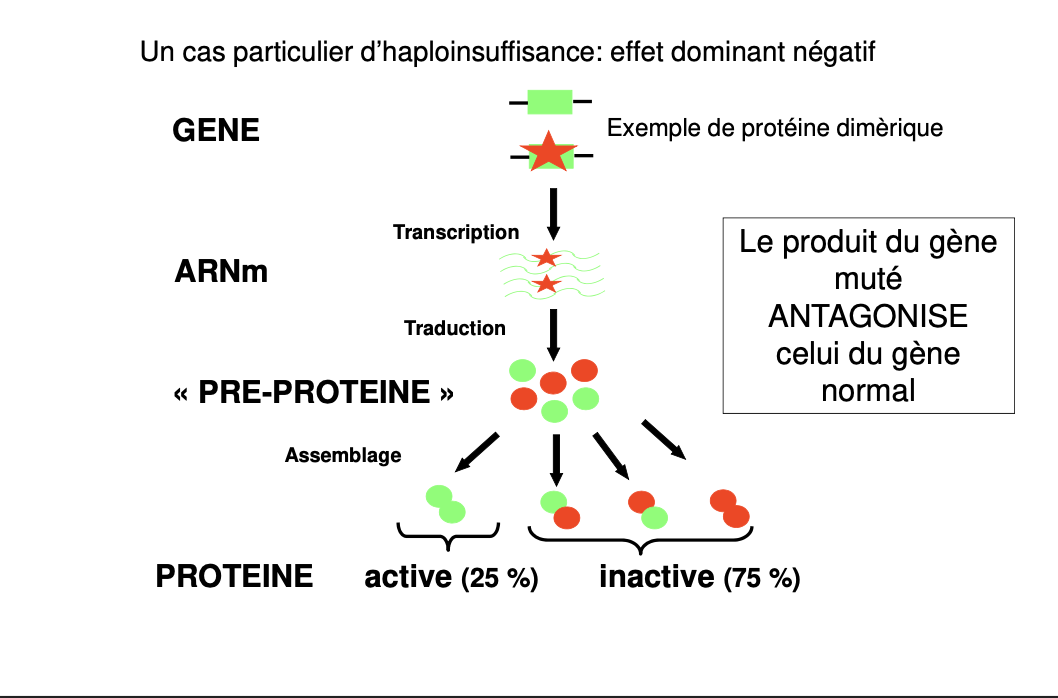
Qu’est-ce que l’haplo-insuffisance (HI) ?
Exemple d’HI avec une protéine trimérique : décrire les 2 variantes « quantité↓ » et « activité↓ ».
Quels indices cliniques/génétiques orientent vers une maladie dominante par HI ?
Qu’est-ce qu’un effet dominant négatif (DN) ?
Comment distinguer DN vs HI expérimentalement (idées d’arguments/tests) ?
Donner un exemple métabolique : enzyme limitante C et conséquences chez l’hétérozygote vs l’homozygote muté.
Quels types de mutations causent plutôt HI vs DN ?
Quelles pistes thérapeutiques selon HI vs DN ?
Dans ce contexte, que veulent dire « quantité de protéine ↓ » vs « activité de protéine ↓ » ?
Perte de fonction
Diminution ou abolition de la fonction du produit génique : soit moins de protéine produite/stable, soit protéine présente mais moins/plus du tout active.Haplo-insuffisance (HI)
Un seul allèle fonctionnel ne suffit pas à atteindre le seuil d’activité requis pour un phénotype normal → l’hétérozygote est malade.Trimère — deux variantes d’HI
HI-quantité : une mutation abolit la transcription/synthèse d’une sous-unité → on assemble moins de trimères fonctionnels → activité totale < seuil.
HI-activité : toutes les sous-unités sont produites, mais la mutation rend une sous-unité inactive → même nombre de trimères, activité spécifique ↓ → activité totale < seuil.
Indices pour HI dominante
Phénotype chez hétérozygote (dominant).
Corrélation au dosage (seuil) ; les homozygotes souvent plus sévères/ létaux.
Mutations souvent tronquantes/perte d’expression ; pas de forme mutante toxique détectable.
Dominant négatif (DN)
La protéine mutée s’assemble avec la protéine normale (complexe multimérique) et sabote le complexe → la présence de l’allèle muté inhibe la fonction de l’allèle sain (effet « poison »).Distinguer DN vs HI (pistes)
Co-expression WT+mutant en cellules : si la co-expression abaisse l’activité en-dessous de la simple demi-dose WT → DN.
Surexprimer la WT : si la surexpression rescue → plutôt HI ; si non-rescue → DN.
Nature des mutations : DN souvent missense conservant l’assemblage ; HI souvent nonsense/frameshift avec NMD (ARNm dégradé).
Dominance de la sévérité : DN peut être très sévère déjà chez l’hétérozygote.
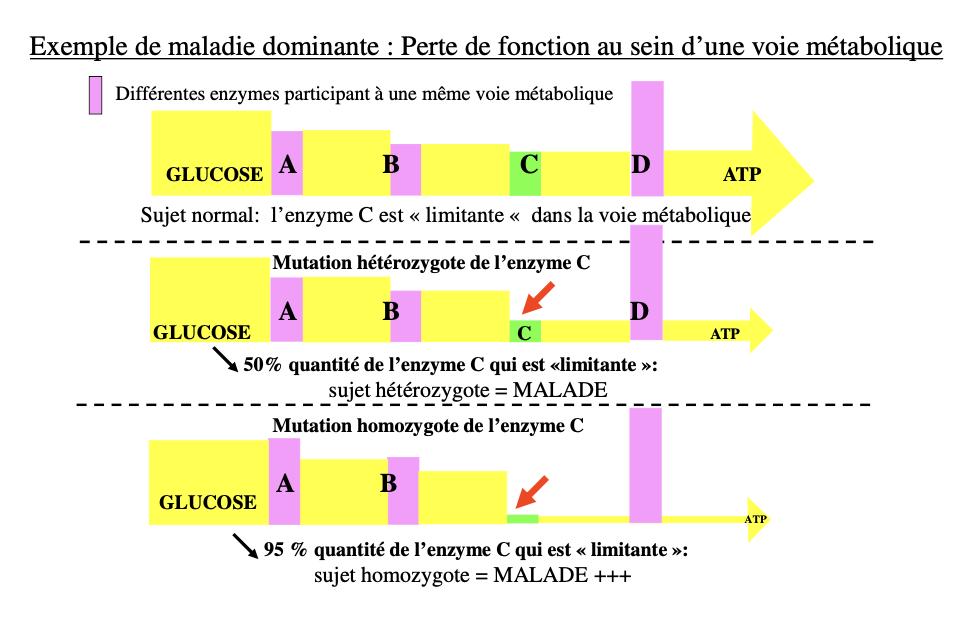
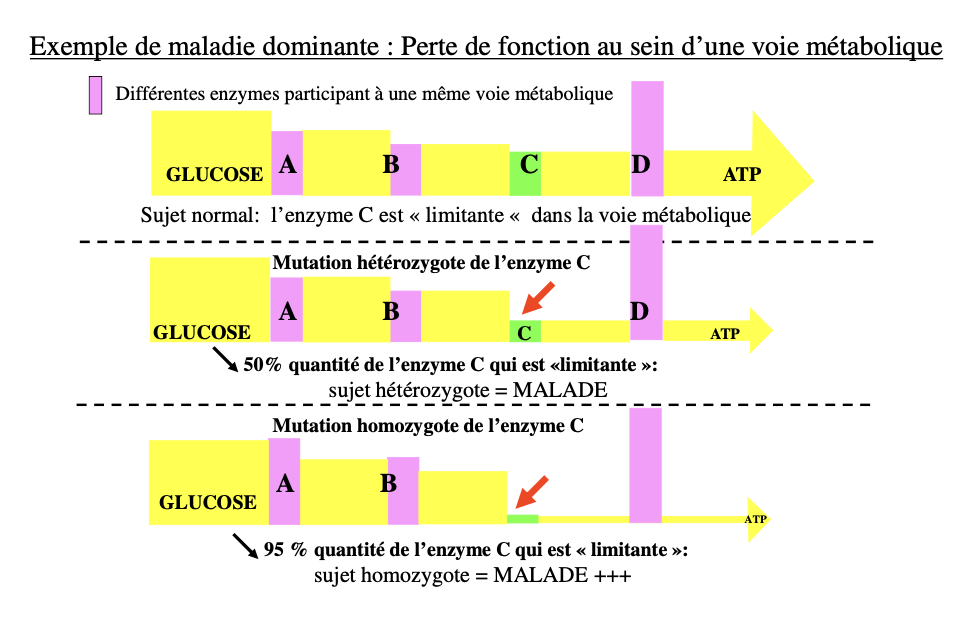
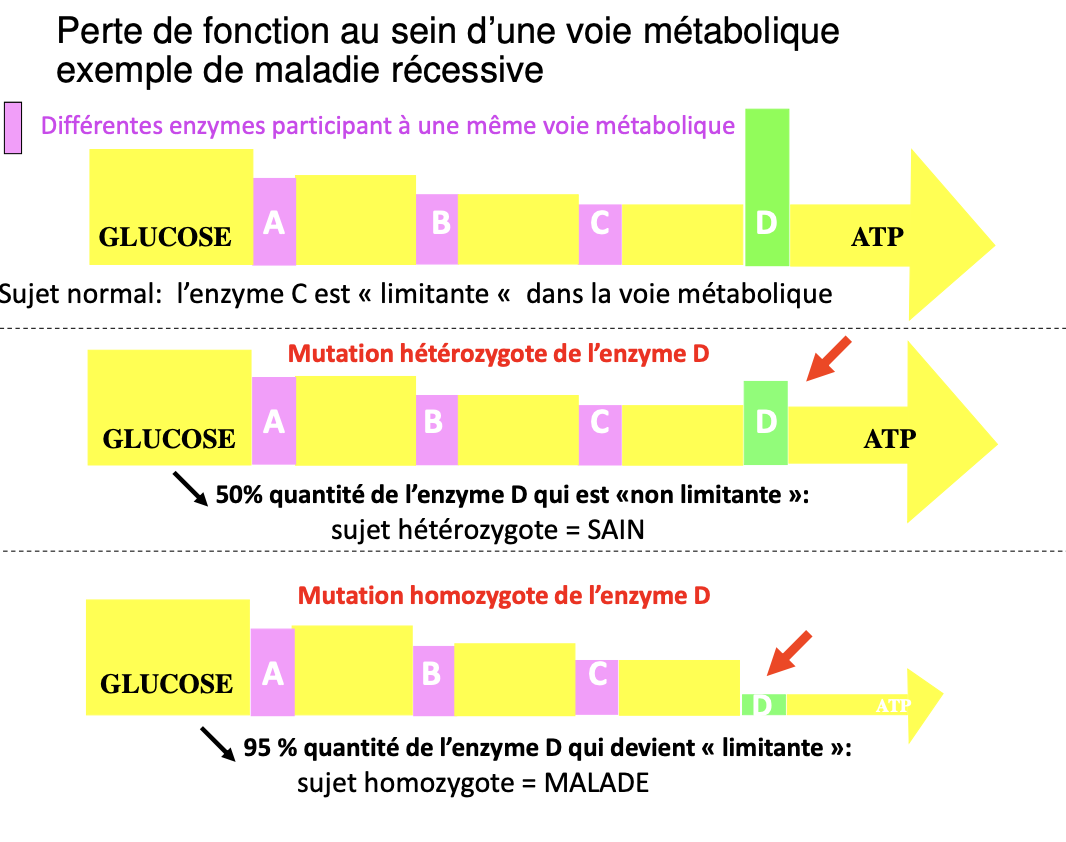
Voie métabolique — enzyme C limitante
Normal : flux glucose → A→B→C (limitante)→D→ATP.
Hétérozygote perte de C : ~50 % d’enzyme C → flux < seuil → malade.
Homozygote : ~5–10 % (ou 0) d’enzyme C → malade ++ / potentiellement létal.
Types de mutations
HI : nonsense, frameshift, délétions, splice létal → absence de protéine (NMD) ou instabilité.
DN : missense structurales qui gardent l’interaction (domaine d’assemblage intact) mais inactivent le site actif/conformation.
Thérapeutique
HI : augmenter la dose/fonction (thérapie génique, correcteurs de repliement, stimulation transcriptionnelle, stabilisation).
DN : inhiber la mutante (silencing allèle-spécifique, PROTAC), favoriser WT (correcteurs d’assemblage), parfois édition pour inactiver l’allèle toxique → convertir en HI puis compenser.
Quantité vs activité
Quantité ↓ : moins de molécules fonctionnelles (expression/stabilité).
Activité ↓ : quantité normale de complexe, mais kcat/affinité altérée → activité globale insuffisante.
2) Gain de fonction

Catégorie 1 — Gain de fonction (autosomique dominant) Partie Questions
Que veut dire “gain de fonction” en génétique ?
Donne 3 mécanismes moléculaires typiques.
Comment différencier gain de fonction, dominant négatif et haplo-insuffisance ?
Quels indices cliniques/exam pointent vers un gain de fonction ?
Partie Réponses
Mutation conférant une activité nouvelle ou augmentée qui domine l’allèle normal.
a) Activation constitutive d’un récepteur/kinase ; b) canal ionique ouvert anormalement ; c) expansion de triplets → protéine toxique.
Gain : activité ↑/nouvelle ; Dominant négatif : l’allèle muté inhibe l’allèle normal ; Haplo-insuffisance : 50 % de produit insuffisant (perte de fonction dominante).
Phénotype malgré dosage à 50 %, récepteurs/kinases hyper-actifs, agrégats toxiques, parfois anticipation.
Catégorie 2 — Achondroplasie (FGFR3) Partie Questions
Gène et rôle physiologique ?
Type de mutation et conséquence cellulaire ?
Mutation “hot spot” la plus fréquente ?
Tableau clinique cardinal (x3) ?
Transmission, part de novo et particularité de l’homozygotie ?
Partie Réponses
FGFR3, récepteur FGF : inhibe la prolifération des chondrocytes.
Mutation activatrice → inhibition excessive du cartilage de croissance.
G380R (c.1138G>A/C).
Petite taille disproportionnée, raccourcissement des os longs, macrocrânie (trident hand, tronc relatif OK, QI normal).
AD, ~80 % de novo, homozygote léthal (souvent néonatal), âge paternel ↑.
Catégorie 3 — Chorée de Huntington (HTT, expansion CAG) Partie Questions
Anomalie moléculaire et gène ?
Seuils de répétitions CAG (normal → complet) ?
Phénomène d’anticipation : par quel parent surtout ?
Triade clinique typique ?
Base physiopath : pourquoi les neurones meurent ?
Partie Réponses
Expansion CAG intragénique du gène HTT → polyglutamine allongée.
≤26 normal ; 27–35 intermédiaire ; 36–39 pénétrance réduite ; ≥40 pénétrance complète.
Anticipation surtout paternelle (expansion à la spermatogenèse).
Mouvements choréiques, troubles psychiatriques, déclin cognitif (30–50 ans).
Misfolding → agrégats protéiques → neurodégénérescence (striatum/cortex).
Catégorie 4 — Comparaisons & “phrases d’exam” Partie Questions
Quelle phrase “code” te fait penser à l’achondroplasie ?
Quelle phrase “code” te fait penser à Huntington ?
Quel mécanisme évoquer si 50 % de produit suffisent normalement mais le phénotype persiste ?
Partie Réponses
“Mutation activatrice d’un récepteur inhibiteur de croissance → retard de croissance.”
“AD + anticipation + CAG (polyQ) → Huntington.”
Gain de fonction ou dominant négatif (pas une simple haplo-insuffisance).
Catégorie 5 — À retenir (chiffres & red flags) Partie Questions
Pour l’achondroplasie : % de novo ? mutation clé ?
Pour Huntington : seuil de pénétrance complète ? parentalité de l’anticipation ?
Qu’indique une homozygotie létale dans une maladie AD ?
Partie Réponses
~80 % de novo, FGFR3 G380R.
≥40 CAG, anticipation paternelle.
Fort gain de fonction délétère (toxicité dose-dépendante).
2 Heredite mendelienne
-automisomique (recessive)
Qu’est-ce qu’un caractère autosomique récessif (AR) ?
Quels indices de mode AR sur un arbre généalogique ?
Quelles probabilités pour un couple porteur × porteur (A/a × A/a) ?
Quel risque pour a/a × A/A ?
Quel risque pour a/a × A/a ?
Qu’est-ce que la pseudodominance et dans quel contexte survient-elle ?
Citer des exemples de maladies AR fréquentes.
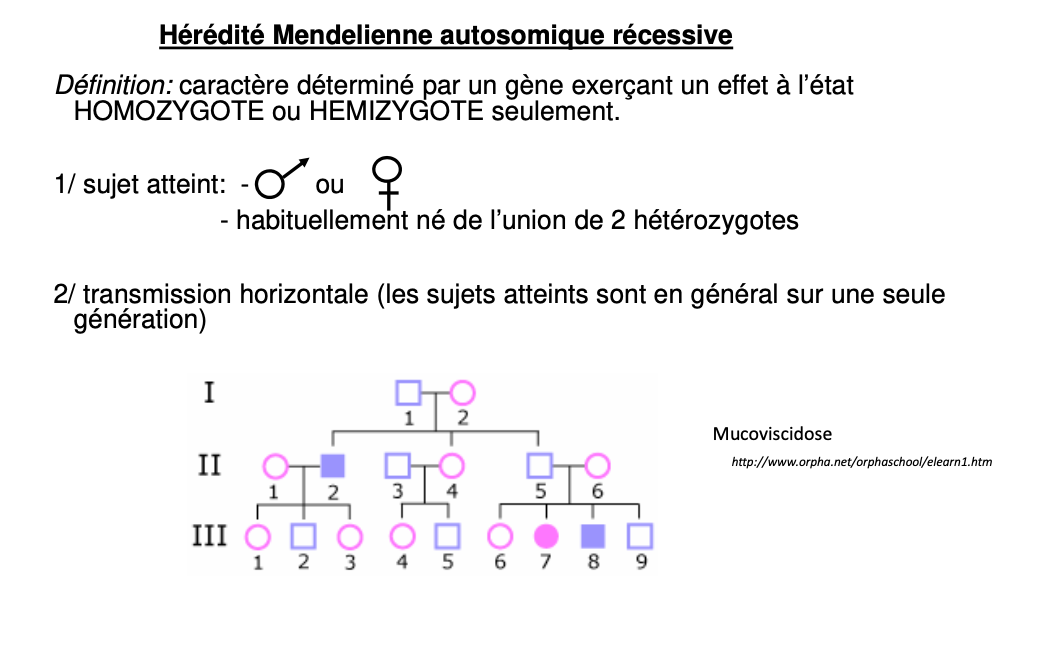

Maladie/trait exprimé uniquement chez l’homozygote muté (a/a) ou hémizygote (gène sur X chez garçon) ; les hétérozygotes (A/a) sont porteurs sains.
Deux sexes touchés, transmission horizontale (atteints surtout parmi frères/sœurs), parents généralement sains mais porteurs, consanguinité augmentant le risque.
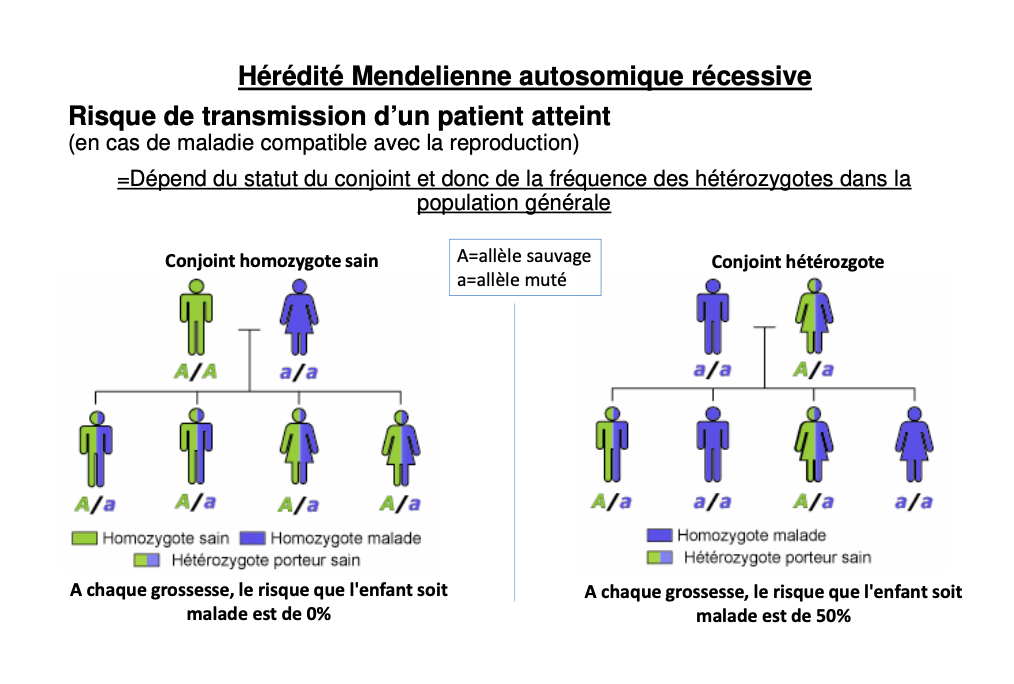
25 % malade (a/a) ; 50 % porteur sain (A/a) ; 25 % sain non porteur (A/A).
0 % malade, 100 % porteurs (A/a).
50 % malade (a/a) ; 50 % porteur (A/a).
Apparence dominante d’une maladie AR quand la fréquence des porteurs est élevée : un patient a/a a souvent un conjoint A/a ⇒ ≈50 % d’enfants atteints ; peut faire croire à une atteinte sur plusieurs générations (ex. drépanocytose).
Mucoviscidose, drépanocytose, phénylcétonurie, thalassémies, etc.

HÉRÉDITÉ MENDELIENNE AUTOSOMIQUE RÉCESSIVE — DÉPISTAGE DES HÉTÉROZYGOTES Questions
Dans quels cas peut-on dépister les hétérozygotes en population générale ?
Quels exemples illustrent un phénotype mesurable ou détectable chez les hétérozygotes ?
Dans quel cas le dépistage repose uniquement sur l’analyse du génotype ?
Que peut-on calculer lorsqu’aucun dépistage direct n’est possible ?

Cas de dépistage possible :
Si le phénotype des hétérozygotes est modifié (caractère mesurable ou détectable).
Si le génotype est identifiable (mutations prévalentes connues dans la population, analysées par l’ADN).
Exemples phénotypiques :
Caractère mesurable : activité enzymatique réduite à 50 % chez un hétérozygote comparé à un homozygote sain.
Caractère détectable : anomalie de l’audiogramme pour un gène de surdité.
Dépistage par génotype :
Lorsque la population générale présente des mutations fréquentes et connues, il est possible de les identifier par analyse moléculaire de l’ADN.
En absence de dépistage direct :
On ne peut fournir qu’une probabilité d’atteinte de la descendance, calculée à partir de la fréquence des hétérozygotes via la loi de Hardy-Weinberg.
HÉRÉDITÉ MENDELIENNE AUTOSOMIQUE RÉCESSIVE & LOI DE HARDY-WEINBERG Questions Définitions & conditions
Quelles sont les conditions nécessaires pour que la loi de Hardy-Weinberg s’applique ?
Quelles sont les fréquences des génotypes possibles selon le modèle de Hardy-Weinberg ?
Application pratique
Si la fréquence d’une maladie autosomique récessive est connue, comment calcule-t-on la fréquence de l’allèle muté ?
Comment calcule-t-on la fréquence des hétérozygotes dans une population selon Hardy-Weinberg ?
Applique : si la mucoviscidose a une prévalence de 1/4500, quelle est la fréquence approximative des hétérozygotes porteurs de la mutation dans la population ?


Définitions & conditions
Conditions d’application de Hardy-Weinberg :
Les mariages doivent être contractés au hasard (panmixie).
La population doit être viable et féconde.
Absence de mutation, migration et sélection.
👉 Ces conditions garantissent une stabilité des fréquences alléliques au cours des générations.
Fréquences génotypiques selon Hardy-Weinberg (p + q = 1) :
f(AA) = p² (homozygotes normaux)
f(aa) = q² (homozygotes atteints)
f(Aa) = 2pq (hétérozygotes porteurs)
Application pratique
Fréquence de l’allèle muté (q) :
Si une maladie autosomique récessive a une fréquence q² = prévalence, alors
q = √(q²)Fréquence des hétérozygotes :
Elle est donnée par la formule :
f(Aa)=2pq
Avec p ≈ 1 lorsque q est petit.Exemple mucoviscidose :
Prévalence : 1/4500 → q² = 1/4500.
Donc q = √(1/4500) ≈ 1/66.
Alors fréquence des hétérozygotes : 2pq ≈ 2 × (1) × (1/66) ≈ 1/33.
👉 Environ 1 individu sur 33 est porteur hétérozygote de la mutation.
NOUVEAUUUUUUUUU
Partie Questions
Quelles sont les bases moléculaires des maladies autosomiques récessives ?
Pourquoi s’agit-il toujours d’une perte de fonction et non d’un gain de fonction ?
Que se passe-t-il au niveau de la quantité ou de l’activité de la protéine mutée ?
Pourquoi les hétérozygotes simples sont-ils cliniquement sains ?
Que signifie l’hétérogénéité allélique ?
Pourquoi certaines mutations récessives sont-elles dites « prévalentes » ?
Quels sont les mécanismes expliquant la fréquence de certaines mutations récessives dans la population ?
Donner des exemples de maladies autosomiques récessives avec leur avantage sélectif associé.

Bases moléculaires : Les maladies autosomiques récessives sont presque toujours liées à des mutations qui entraînent une perte de fonction de la protéine codée.
Perte et non gain de fonction : Un gain de fonction affecterait aussi les hétérozygotes simples, qui seraient malades. Or, dans les maladies autosomiques récessives, seuls les homozygotes sont atteints → il s’agit donc toujours d’une perte de fonction.
Protéine mutée : La mutation provoque une diminution de la quantité ou de l’activité enzymatique/fonctionnelle de la protéine.
Hétérozygotes sains : Chez eux, 50 % d’activité protéique est suffisante pour maintenir la voie métabolique ou la fonction cellulaire concernée dans des conditions normales.
Hétérogénéité allélique : La maladie peut être causée par un grand nombre de mutations différentes réparties sur l’ensemble du gène (y compris dans les régions non traduites), chacune induisant une perte de fonction.
Mutations prévalentes : Certaines maladies récessives sont associées à un petit nombre de mutations fréquentes dans la population, appelées mutations prévalentes.
Mécanismes expliquant la prévalence :
Effet fondateur : propagation d’une mutation unique dans une population restreinte à l’origine.
Avantage sélectif : les hétérozygotes porteurs ont une meilleure survie face à certaines conditions environnementales (ex : résistance à une infection, meilleure adaptation physiologique).
Exemples :
Drépanocytose (mutation Glu6Val du gène de la β-globine) → protection contre le paludisme chez les hétérozygotes.
Mucoviscidose (mutation F508del du gène CFTR) → protection contre la déshydratation par pertes excessives d’eau.
im occ
Quel est le risque pour un couple d’hétérozygotes simples d’avoir un enfant atteint ?
Quel est le risque pour ce couple d’avoir un enfant sain ?
Quel est le risque pour un enfant sain issu d’un couple d’hétérozygotes simples d’être lui-même hétérozygote ?
Comment peut-on dépister les hétérozygotes dans la population générale ?
De quoi dépend la probabilité pour un individu de la population générale d’être hétérozygote ?
Comment peut-on évaluer la probabilité d’hétérozygotie dans une population ?
Quel est l’impact de la consanguinité parentale sur le risque de maladie récessive ?
Quelle est la base moléculaire d’une maladie autosomique récessive ?
Qu’entend-on par hétérogénéité allélique dans ces maladies ?
Pourquoi certaines mutations autosomiques récessives persistent-elles dans la population ?

2 Heredite mendelienne
liee au sexe
dominante (DLX)
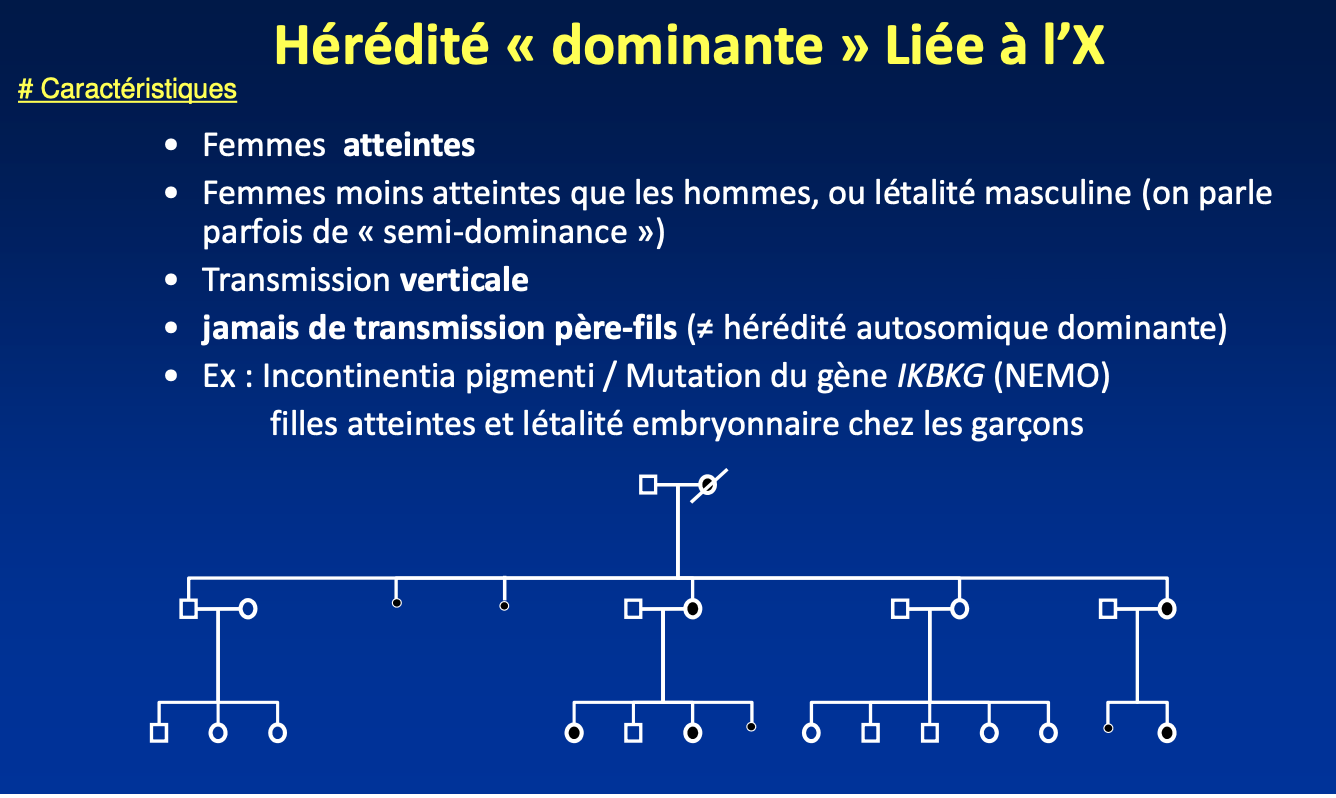
Qu’est-ce que l’hérédité dominante liée à l’X (DLX) ?
Quelle est la différence entre les hommes et les femmes face à une maladie DLX ?
Quelles sont les caractéristiques de transmission d’une maladie DLX ?
Pourquoi n’existe-t-il jamais de transmission père-fils dans ce mode d’hérédité ?
Donner un exemple de maladie dominante liée à l’X et ses particularités.

Qu’est-ce que l’hérédité dominante liée à l’X (DLX) ?
C’est une forme d’hérédité où les caractères pathologiques sont portés par des gènes situés sur le chromosome X. Elle est dite dominante car une seule copie mutée du gène (chez l’homme ou la femme) suffit à entraîner la maladie.
Quelle est la différence entre les hommes et les femmes face à une maladie DLX ?
Hommes (XY) : ils sont hémizygotes pour le chromosome X. Ainsi, s’ils portent une mutation sur leur unique X, ils expriment pleinement la maladie, parfois de façon sévère ou létale.
Femmes (XX) : elles possèdent deux X et donc deux copies du gène. Elles peuvent être atteintes, mais souvent moins sévèrement que les hommes, car elles conservent une copie fonctionnelle. On parle parfois de semi-dominance.
Quelles sont les caractéristiques de transmission d’une maladie DLX ?
Transmission verticale : la maladie se retrouve de génération en génération.
Jamais de transmission père-fils : car les pères transmettent leur chromosome Y à leurs fils.
Les filles d’un père atteint seront toutes touchées (car il transmet obligatoirement son X muté).
Les mères hétérozygotes transmettent la mutation à 50 % de leurs enfants (filles et garçons).
Pourquoi n’existe-t-il jamais de transmission père-fils dans ce mode d’hérédité ?
Parce que le père transmet toujours son chromosome Y à ses fils, jamais son chromosome X. Ainsi, un gène muté porté sur l’X paternel ne peut pas être transmis directement à un garçon.
Donner un exemple de maladie dominante liée à l’X et ses particularités.
Incontinentia pigmenti (IP), due à une mutation du gène IKBKG (NEMO).
Particularités :
Les femmes hétérozygotes sont atteintes (atteintes cutanées, dentaires, neurologiques).
La mutation est souvent létale chez les garçons pendant le développement embryonnaire → seules les filles naissent avec la maladie.
2 Heredite mendelienne
liee au sexe
dominante (DLX)
Qu’est-ce qu’une hérédité dominante liée à l’X (DLX) ?
Quelles sont ses caractéristiques principales ?
Quels sont les modes de transmission possibles ?
Que se passe-t-il dans le cas particulier de parents indemnes avec enfant atteint ?

1. Définition de l’hérédité dominante liée à l’X (DLX)
Liée à l’X : les gènes responsables sont localisés sur le chromosome X.
Dominante : le phénotype s’exprime même en présence d’un seul allèle muté, aussi bien chez l’homme (hémizygote) que chez la femme (hétérozygote).
Les hommes n’ayant qu’un seul chromosome X sont hémizygotes : toute mutation dominante sur l’X se manifeste chez eux.
Les femmes ont deux chromosomes X : elles peuvent être hétérozygotes (atteintes) ou homozygotes (souvent létal).
2. Caractéristiques cliniques et transmission
Les femmes sont atteintes, souvent moins sévèrement que les hommes (effet mosaïque lié à l’inactivation de l’X).
Les hommes sont plus sévèrement atteints ; certaines mutations sont létales chez eux (ex. semi-dominance ou létalité embryonnaire).
Transmission verticale : la maladie se retrouve de génération en génération.
Jamais de transmission père-fils, puisque le père transmet son chromosome Y à ses fils.
Exemple : Incontinentia pigmenti (IKBKG/NEMO) → filles atteintes, garçons létaux.
3. Modes de transmission
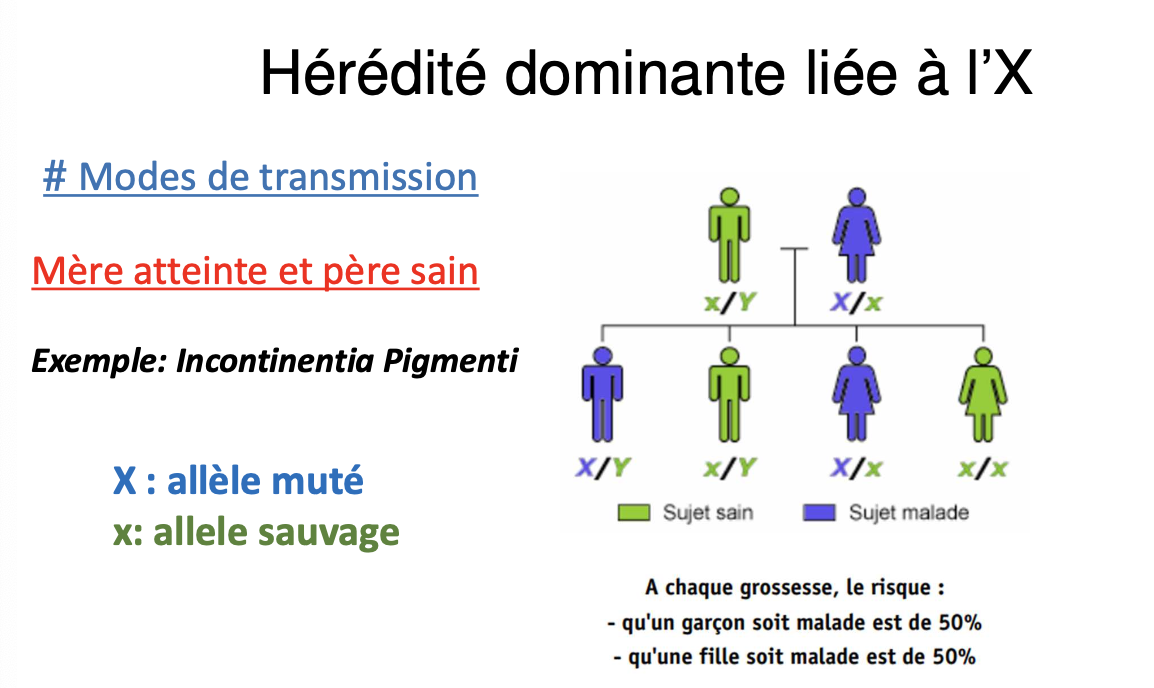
Mère atteinte et père sain :
50 % de risque qu’un garçon soit atteint.
50 % de risque qu’une fille soit atteinte.
Exemple : Incontinentia Pigmenti.
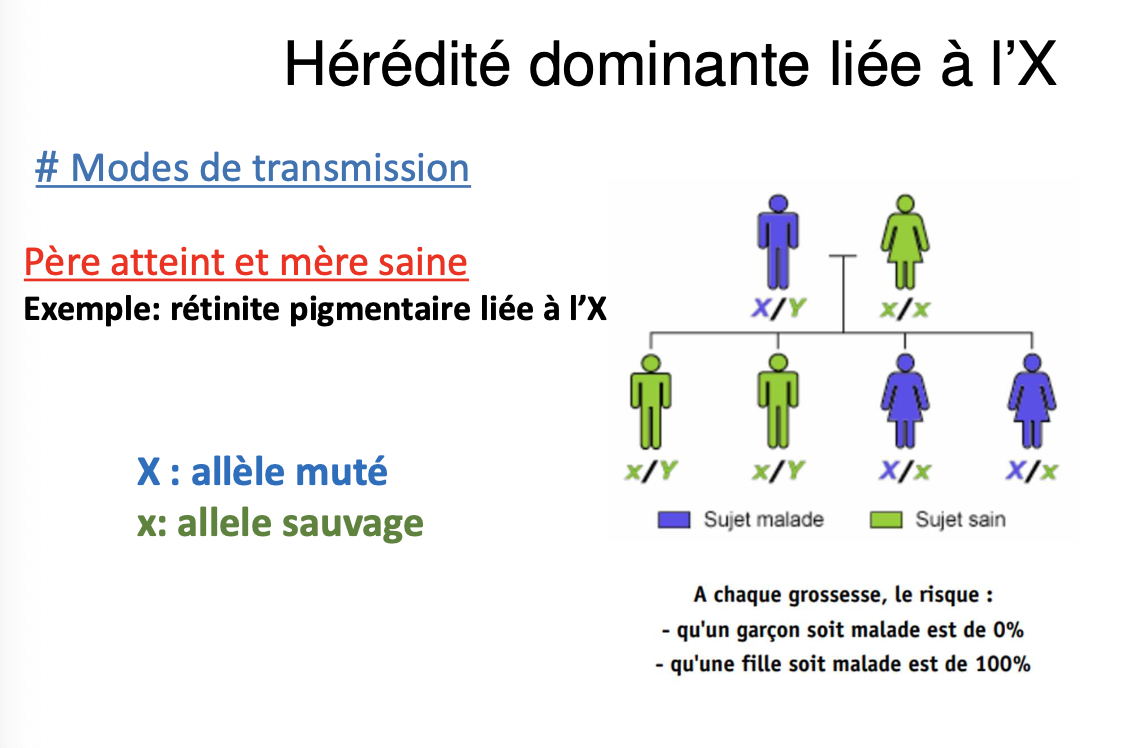
Père atteint et mère saine :
Aucun fils n’est atteint (père transmet Y).
Toutes les filles sont atteintes (100 %).
Exemple : rétinite pigmentaire liée à l’X.
Cas particulier : parents indemnes avec enfant atteint :
Résulte d’une mutation de novo ou néomutation dans une cellule germinale (spermatozoïde ou ovocyte).
Risque de récurrence très faible, sauf en cas de mosaïcisme germinal.
Analyser l’arbre et donner les risques pour la descendance.

2 Heredite mendelienne
liee au sexe
dominante (DLX)
Qu’est-ce qu’on entend par hérédité dominante liée à l’X (DLX) ?
Pourquoi les hommes sont-ils dits hémizygotes pour les gènes du chromosome X ?
Quelles sont les principales caractéristiques cliniques et de transmission de l’hérédité dominante liée à l’X ?
Quels sont les modes de transmission possibles (mère atteinte/père sain, père atteint/mère saine, parents indemnes) et leurs conséquences sur la descendance ?
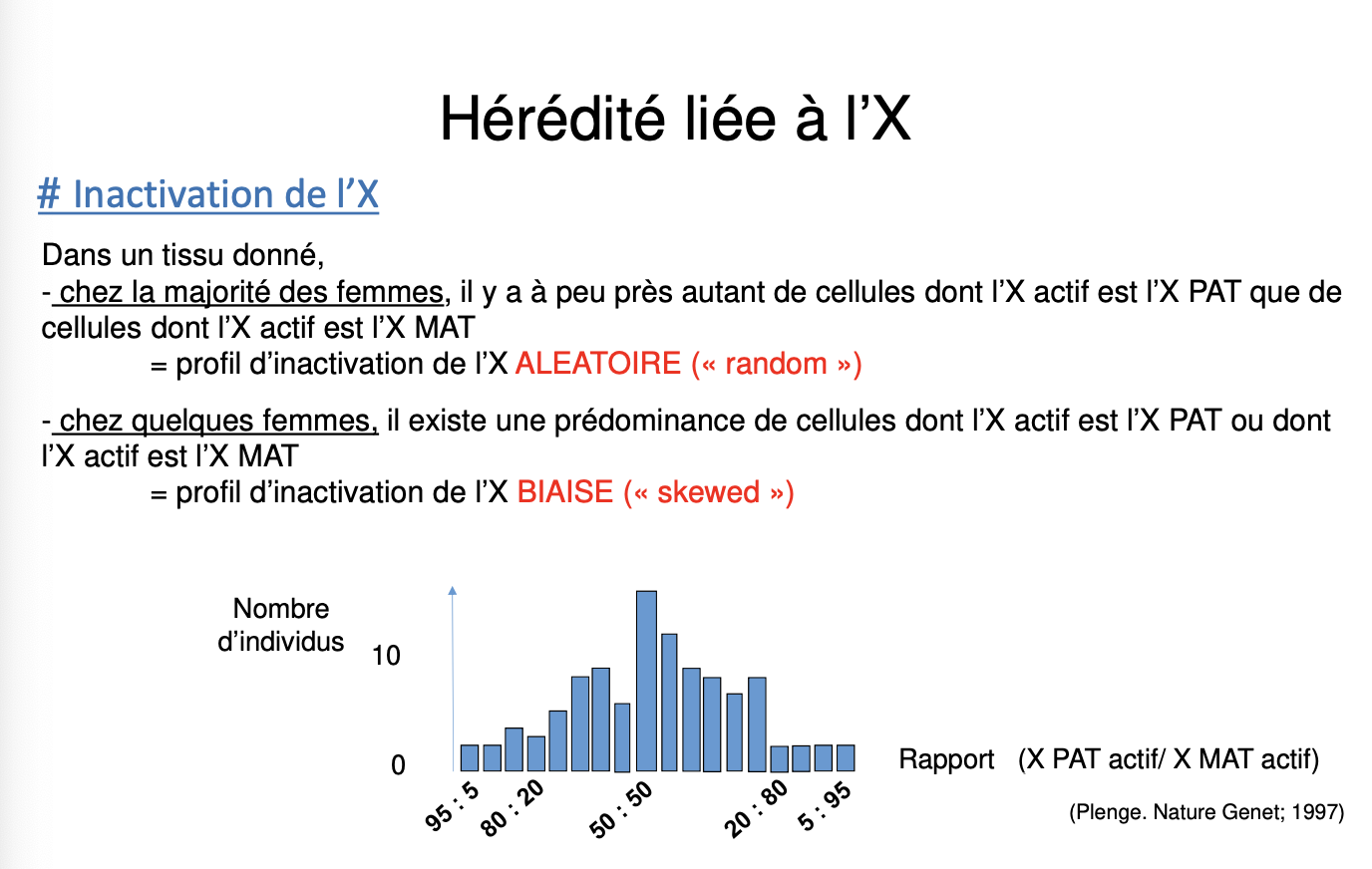
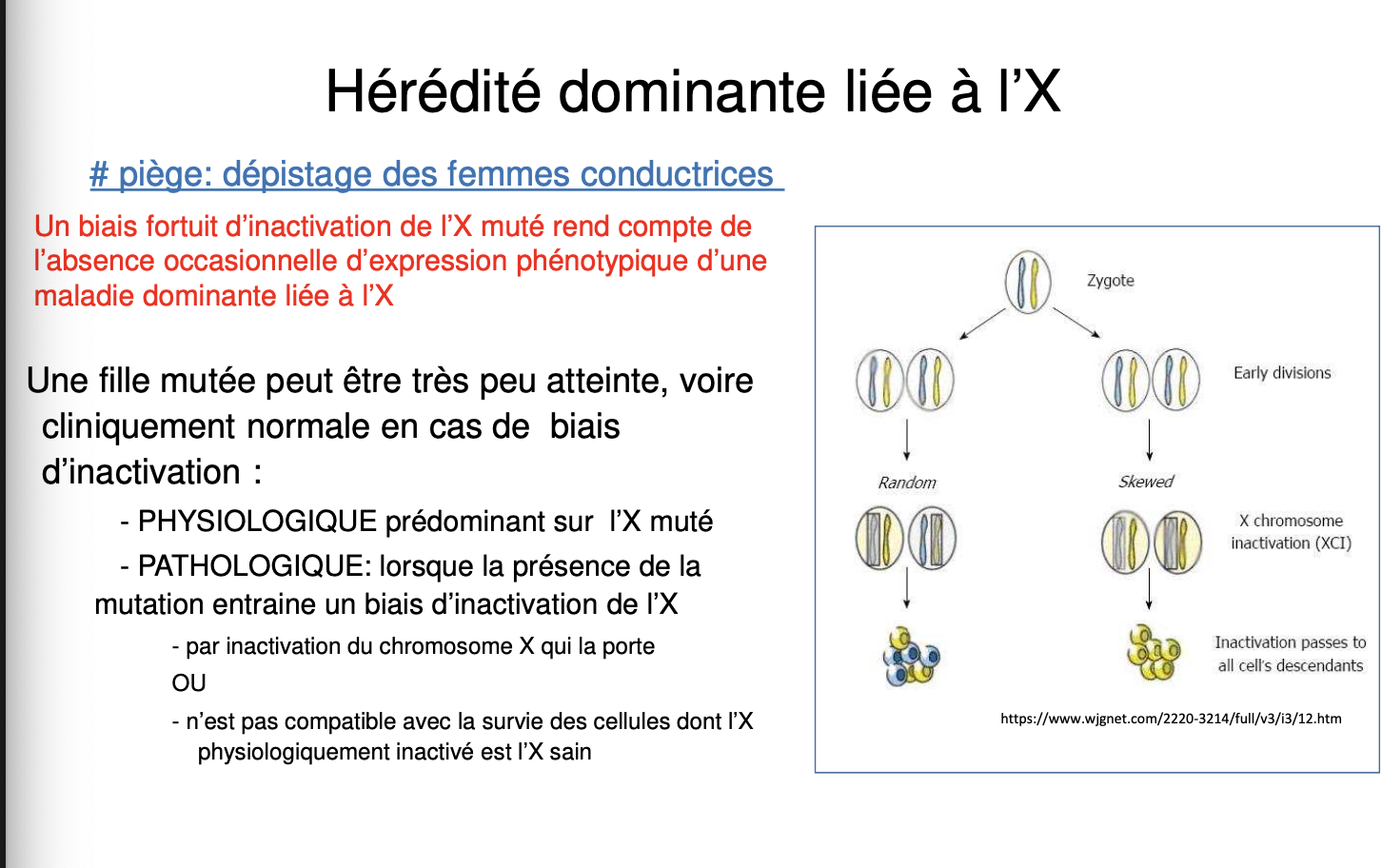
Qu’est-ce que l’inactivation du chromosome X et quelles en sont les propriétés principales (précocité, aléatoire, clonale, définitive) ?
Quelle est la différence entre une inactivation de l’X aléatoire et une inactivation biaisée (skewed) ?
Quel rôle joue le gène XIST dans l’inactivation de l’X ?
Pourquoi l’inactivation biaisée peut-elle compliquer le dépistage des femmes conductrices pour une maladie DLX ?
Quelles sont les différences entre un biais physiologique et un biais pathologique d’inactivation ?
Donner des exemples de maladies à transmission dominante liée à l’X.


Définition :
Une hérédité dominante liée à l’X (DLX) correspond à des maladies dont le gène responsable est situé sur le chromosome X et dont le caractère pathologique s’exprime en présence d’un seul allèle muté.Hommes hémizygotes :
Les individus de sexe masculin ne possèdent qu’un seul chromosome X (XY), donc un seul exemplaire de chaque gène porté par ce chromosome. Toute mutation sur cet X unique s’exprime directement → hémizygotie.Caractéristiques :
Femmes atteintes (car deux X, l’un pouvant être muté).
Atteinte souvent moins sévère chez les femmes que chez les hommes (effet de l’inactivation de l’X).
Transmission verticale (de génération en génération).
Jamais de transmission père-fils (le père transmet son Y aux fils).
Exemple typique : Incontinentia pigmenti (IKBKG/NEMO).
Modes de transmission :
Mère atteinte / père sain → 50 % de risque pour un enfant, qu’il soit garçon ou fille, d’être malade.
Père atteint / mère saine → tous les fils sont sains (Y transmis), toutes les filles sont malades (X transmis).
Parents indemnes → cas de néomutation (de novo), souvent liée à une mutation gamétique, avec risque de mosaïque germinale (faible mais non nul).
Inactivation de l’X :
Précoce : survient dès J3–J6 après fécondation.
Aléatoire : l’X paternel ou maternel peut être inactivé.
Clonale : toutes les cellules filles conservent le même X inactivé.
Définitive : sauf pour les cellules souches germinales (réactivation possible).
Aléatoire vs biaisée :
Aléatoire (random) → environ 50 % des cellules expriment l’X paternel, 50 % l’X maternel.
Biaisée (skewed) → prédominance d’un X actif (soit paternel soit maternel), pouvant modifier l’expression clinique de la maladie.
Rôle du gène XIST :
Le gène XIST, situé sur l’X inactivé, code un ARN non codant qui recouvre et silence transcriptionnellement ce chromosome.Impact sur le dépistage :
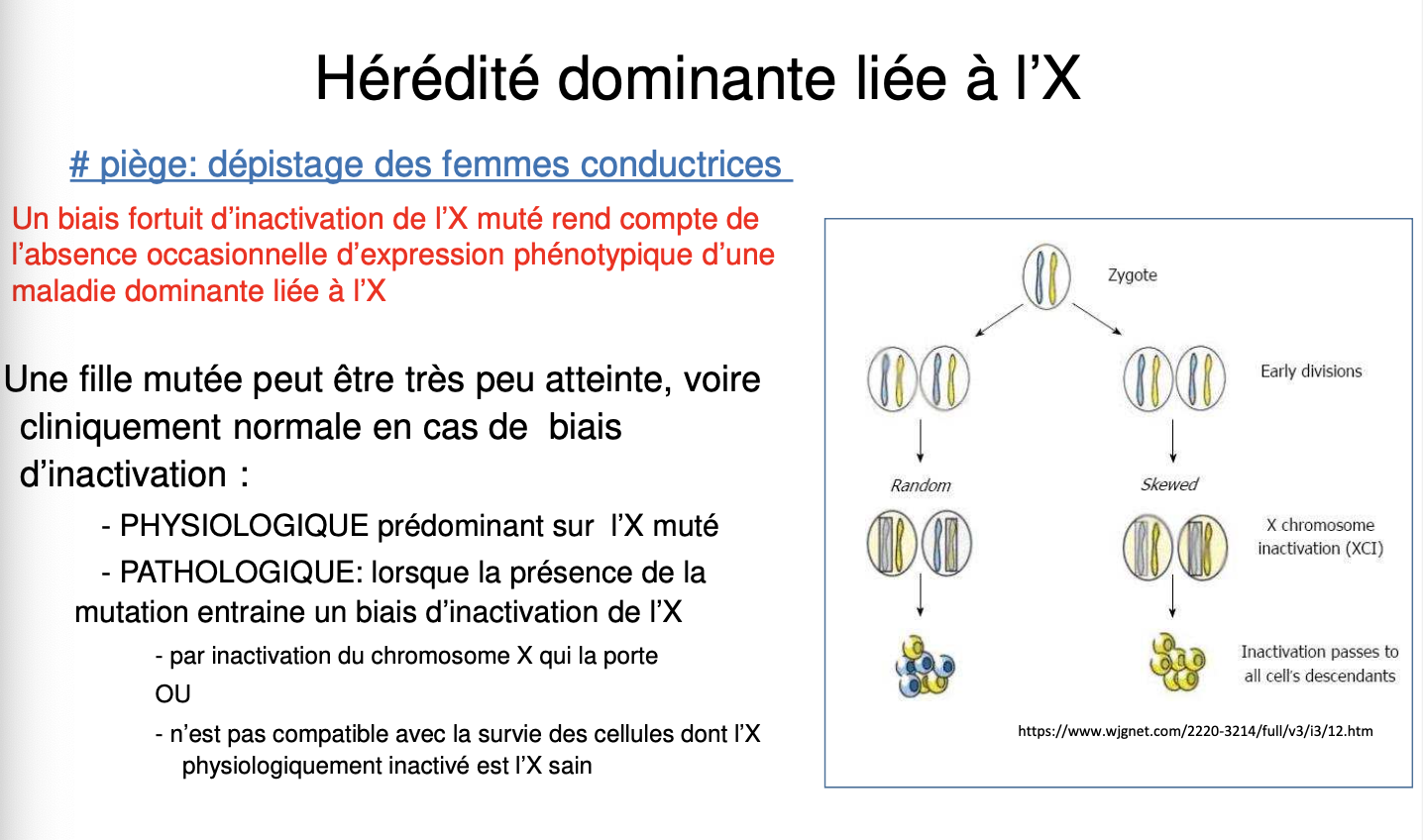
Chez les femmes conductrices, si l’X muté est inactivé préférentiellement, le phénotype peut être atténué voire absent, compliquant le diagnostic.Biais physiologique vs pathologique :
Physiologique : simple hasard d’inactivation donnant une prédominance de l’X sain → atténuation du phénotype.
Pathologique : mutation entraînant une sélection négative contre les cellules portant l’X muté actif → aggravation ou variabilité clinique.
Exemples de maladies DLX :
Incontinentia pigmenti (IKBKG/NEMO).
Rétinite pigmentaire liée à l’X.
Autres maladies rares : syndrome de Rett (MECP2), hypophosphatémie liée à l’X.
IM OCC
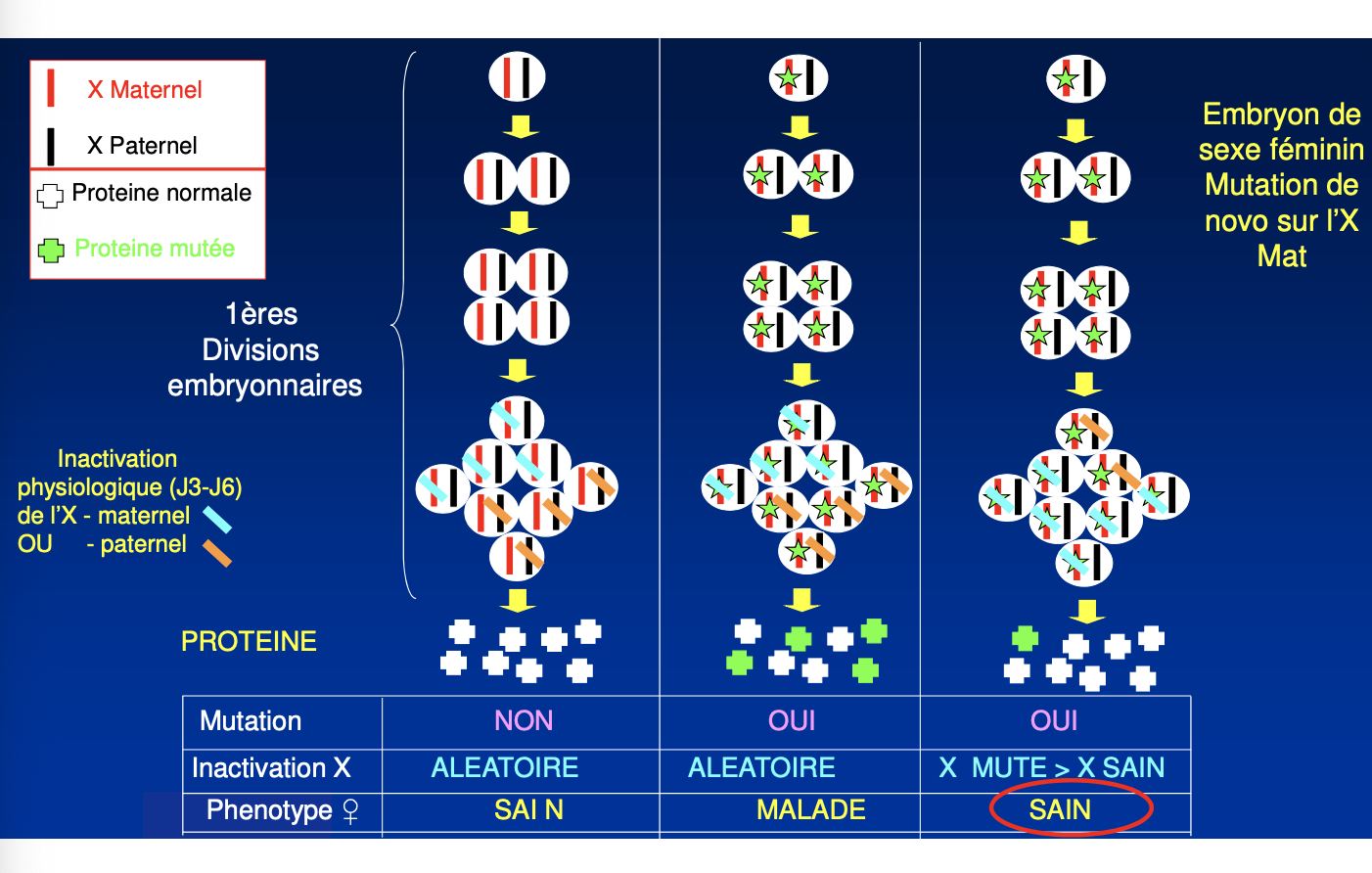
2 Heredite mendelienne
liee au sexe
dominante (DLX)
Qu’est-ce qu’une maladie dominante liée à l’X ?
Quelles sont les caractéristiques cliniques et de transmission des maladies DLX ?
Quels sont les modes de transmission des maladies DLX ?
Quel est le rôle de l’inactivation de l’X dans les maladies DLX ?
Comment dépister les femmes conductrices d’une maladie DLX ?

Définition
Une maladie dominante liée à l’X est une pathologie dont les gènes responsables sont situés sur le chromosome X.
Les hommes sont hémizygotes : ils ne possèdent qu’un seul allèle. S’ils portent l’allèle muté, ils sont atteints.
Les femmes possèdent deux chromosomes X : elles peuvent être atteintes si l’un des deux X est muté.
Le caractère dominant signifie que la mutation s’exprime chez les hommes et chez les femmes.
Caractéristiques
Les femmes sont atteintes, mais en général moins sévèrement que les hommes, car elles disposent d’un deuxième chromosome X fonctionnel.
Chez certains hommes, la mutation est létale dès le stade embryonnaire.
Transmission verticale (présente à chaque génération).
Il n’existe jamais de transmission père → fils, car le père transmet son Y aux garçons.
Exemple : Incontinentia pigmenti (mutation du gène IKBKG/NEMO).
Modes de transmission
Mère atteinte et père sain :
50 % des fils atteints
50 % des filles atteintes
Exemple : Incontinentia pigmenti
Père atteint et mère saine :
0 % des fils atteints (père transmet le Y)
100 % des filles atteintes (père transmet son X muté)
Exemple : Rétinite pigmentaire liée à l’X
Parents indemnes :
Apparition d’une mutation de novo (néomutation).
Risque de récurrence ≃ 0 sauf mosaïcisme germinal.
Inactivation de l’X et biais
L’inactivation de l’X est un processus :
Précoce : entre J3 et J6 post-fécondation.
Aléatoire : concerne l’X paternel ou maternel.
Clonal : toutes les cellules filles gardent le même X inactif.
Définitif : sauf pour les cellules souches germinales.
Conséquences cliniques :
Certaines femmes porteuses de la mutation sont peu ou pas atteintes si l’X muté est majoritairement inactivé.
Un biais d’inactivation peut être :
Physiologique : l’X muté est inactivé, ce qui protège.
Pathologique : seule la survie des cellules avec X muté inactivé est possible → biais dans l’expression.
Dépistage des femmes conductrices
Phénotypage : recherche de signes cliniques mineurs (ex. anomalies cutanées légères dans Incontinentia pigmenti).
Génotypage : identification de la mutation dans le cas index, puis recherche chez la mère.
Si mutation absente → néomutation → risque de récidive ≃ 0 (sauf mosaïcisme germinal).
Si mutation présente → risque de récidive 50 %, quel que soit le sexe fœtal.
2 Heredite mendelienne
liee au sexe
dominante (DLX)
Qu’est-ce que l’hérédité dominante liée à l’X (DLX) ?
Quelles sont les caractéristiques cliniques et génétiques de l’hérédité DLX ?
Quels sont les modes de transmission possibles d’une maladie DLX (mère atteinte/père sain, père atteint/mère saine, parents indemnes) ?
Qu’est-ce que l’inactivation du chromosome X et quelles sont ses propriétés principales (précocité, hasard, caractère clonal, définitif) ?
Quelle est la différence entre une inactivation aléatoire et une inactivation biaisée de l’X ?
En quoi le biais d’inactivation de l’X complique-t-il le dépistage des femmes conductrices ?
Quels sont les pièges diagnostiques liés au phénotype des garçons atteints dans les maladies DLX ?
Quelles sont les bases moléculaires des maladies DLX ?


Définition de l’hérédité DLX
Il s’agit d’une hérédité dont les caractères pathologiques sont portés par le chromosome X et s’expriment de manière dominante, aussi bien chez les hommes (hémizygotes) que chez les femmes (hétérozygotes ou homozygotes).
Caractéristiques cliniques et génétiques
Les femmes sont atteintes, mais souvent moins sévèrement que les hommes, voire la mutation peut être létale chez ces derniers (ex. létalité embryonnaire masculine).
Transmission verticale (de génération en génération).
Jamais de transmission père-fils, car le père transmet son Y au fils.
Exemple : Incontinentia pigmenti (mutation du gène IKBKG/NEMO).
Modes de transmission
Mère atteinte + père sain : chaque grossesse = 50 % de risque pour un fils malade et 50 % pour une fille malade.
Père atteint + mère saine : tous les fils seront sains, mais 100 % des filles seront atteintes.
Parents indemnes : survenue possible d’une néomutation (mutation de novo) dans une gamète. Le risque de récurrence est en principe nul, sauf en cas de mosaïcisme germinal.
Inactivation du chromosome X : propriétés
Précoce : entre J3 et J6 post-fécondation.
Aléatoire : touche indifféremment l’X paternel ou maternel.
Clonale : toutes les cellules filles héritent du même X inactif.
Définitive : sauf pour les cellules souches germinales (où l’X peut être réactivé).
Inactivation aléatoire vs biaisée
Aléatoire (random) : environ 50 % des cellules expriment l’X paternel et 50 % l’X maternel.
Biaisée (skewed) : prédominance d’un X actif (paternel ou maternel), entraînant une inactivation déséquilibrée et des conséquences phénotypiques.
Biais d’inactivation et dépistage des femmes conductrices
Une femme porteuse peut être très peu atteinte, voire cliniquement normale, si l’X muté est majoritairement inactivé. Cela complique :
le dépistage par phénotypage (signes cutanés minimes, expression atténuée).
le génotypage reste l’approche fiable pour identifier la mutation.
Pièges liés au phénotype des garçons atteints
Chez les garçons, les affections DLX sont souvent plus sévères que chez les filles.
Certaines mutations sont létales in utero chez les mâles → absence de naissances masculines atteintes et fausses couches spontanées.
Dans la fratrie, on retrouve surtout des filles atteintes/saines et des garçons sains.
Bases moléculaires
Les maladies DLX résultent d’un gain ou d’une perte de fonction de la protéine codée.
Leur sévérité chez la femme est modulée par le degré d’inactivation de l’X muté :
Si l’X muté est largement inactivé → phénotype discret.
Si l’X sain est majoritairement inactivé → phénotype sévère.
2 Heredite mendelienne
liee au sexe
dominante (DLX)
Quels individus peuvent être atteints d’une maladie dominante liée à l’X (DLX) ?
Quelles sont les origines possibles d’une mutation DLX ?
Quelle est la probabilité d’atteinte dans la descendance d’une femme vectrice ?
Par quels moyens peut-on dépister les femmes vectrices ?
Les femmes vectrices présentent-elles des symptômes ?
Pourquoi une femme vectrice peut-elle paraître saine cliniquement ?
Quelle est la transmission observée dans la descendance d’un homme atteint (transmetteur de sexe masculin) ?
Que se passe-t-il dans les formes sévères de maladies DLX pour les garçons porteurs de la mutation ?
Quelles sont les bases moléculaires des maladies DLX ?
Pourquoi certaines mutations DLX entraînent-elles la mort cellulaire ?

2 Heredite mendelienne
liee au sexe
récessive (RLX)
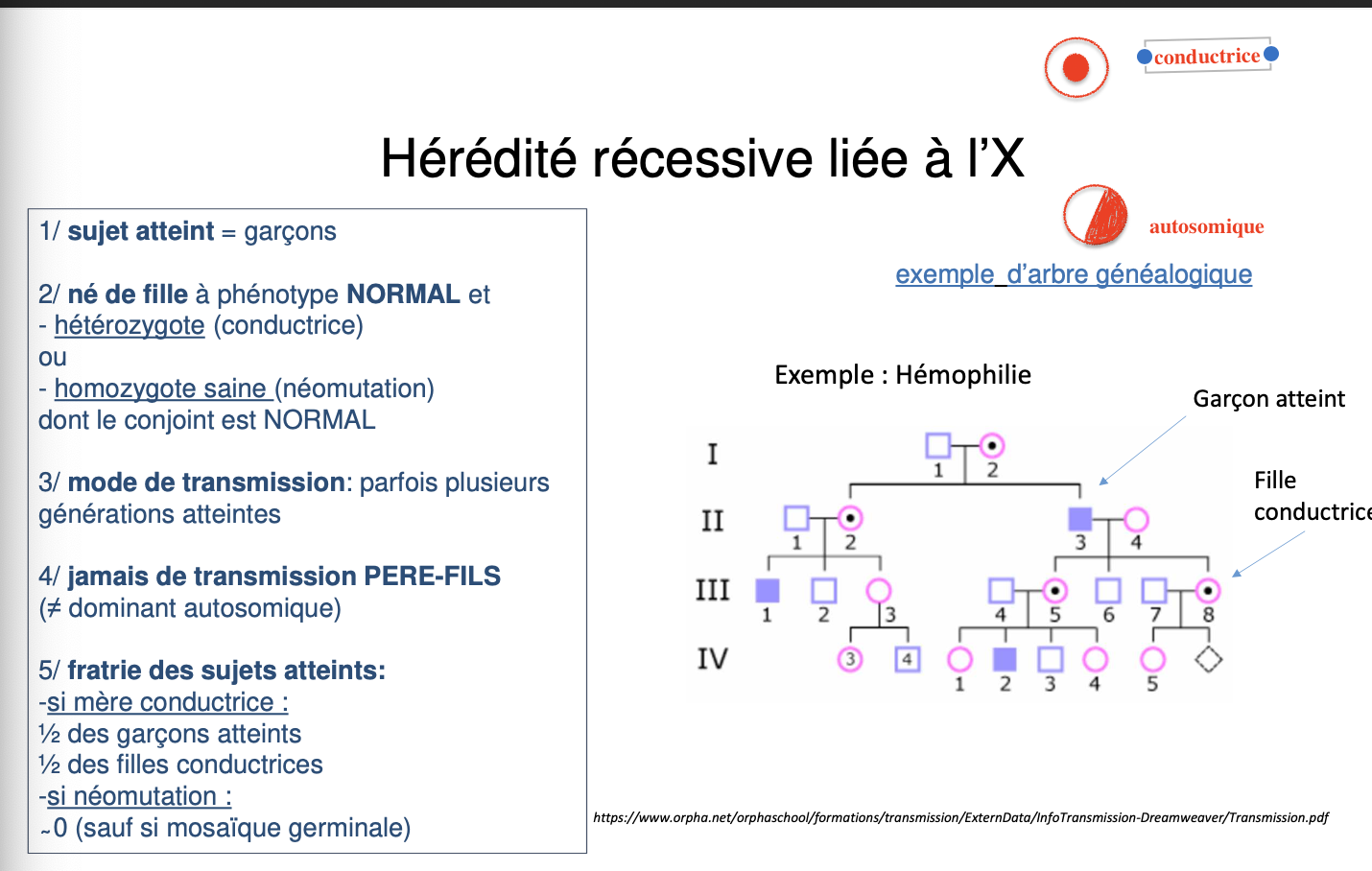
Quelle est la définition d’une hérédité récessive liée à l’X (RLX) ?
Pourquoi les maladies RLX s’expriment-elles surtout chez les garçons ?
Quels sont les risques de transmission si la mère est conductrice (hétérozygote) ?
Quels sont les risques de transmission si le père est atteint ?
Pourquoi parle-t-on d’un risque pour les petits-fils mâles lorsque le père est atteint ?


Définition
L’hérédité récessive liée à l’X correspond à une transmission de caractères pathologiques portés par le chromosome X.
Expression surtout chez les garçons
Les garçons n’ayant qu’un seul chromosome X (ils sont hémizygotes), une mutation récessive portée par ce chromosome s’exprime directement, faute d’allèle normal compensateur.
Transmission mère conductrice (Xx)
À chaque grossesse, le risque est :
50 % d’avoir un fils malade (x/Y),
50 % d’avoir une fille conductrice (Xx).
Transmission père atteint (x/Y)
Un père atteint transmet son chromosome X muté à toutes ses filles, qui seront donc 100 % conductrices (Xx), mais aucun fils ne sera malade car il reçoit le chromosome Y paternel.
Risque pour les petits-fils mâles
Les filles conductrices (obligatoirement hétérozygotes si leur père est atteint) peuvent ensuite transmettre l’allèle muté à leur descendance : leurs fils ont alors 50 % de risque d’être malades, ce qui explique le risque d’atteinte des petits-fils mâles.
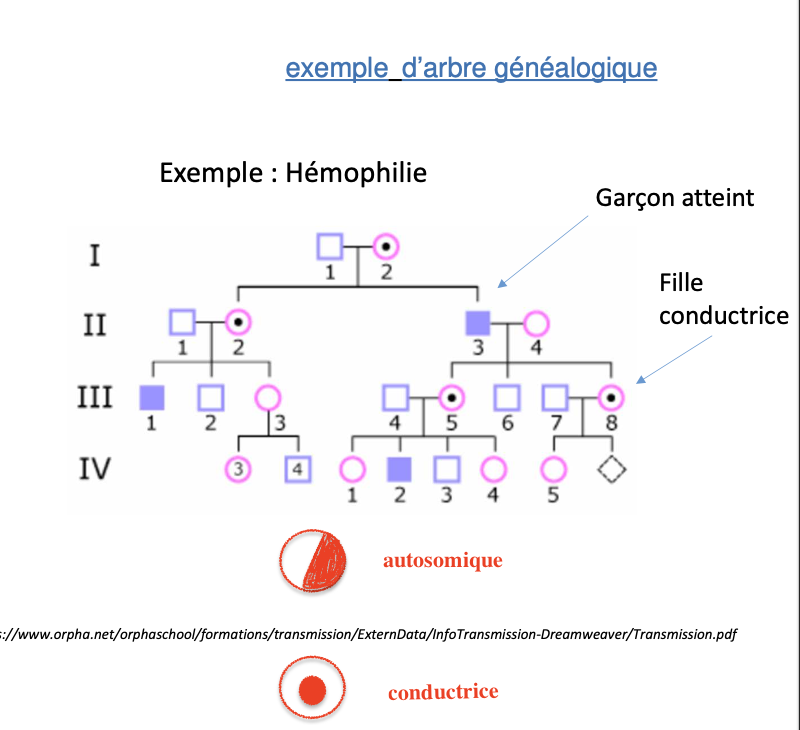
Analyser l’arbre.
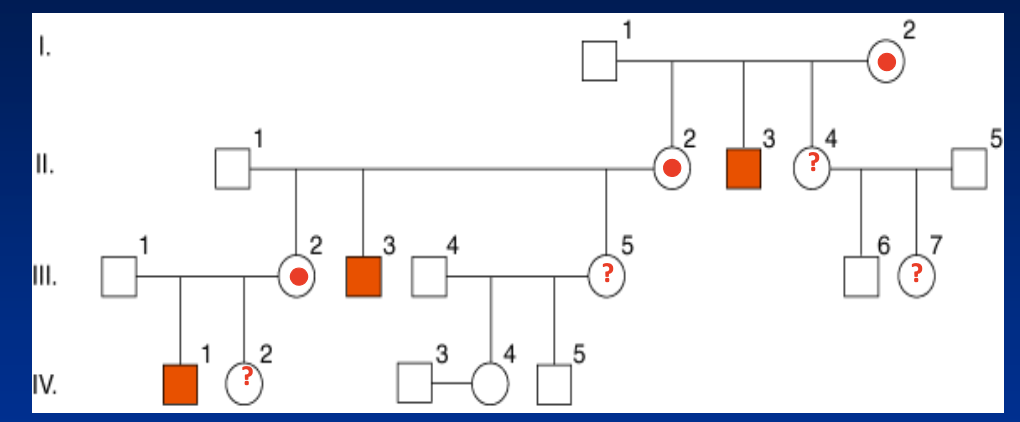
Qu’est-ce que l’hérédité récessive liée à l’X ?
Que se passe-t-il si la mère est conductrice et le père sain ?
Que se passe-t-il si le père est atteint et la mère saine ?
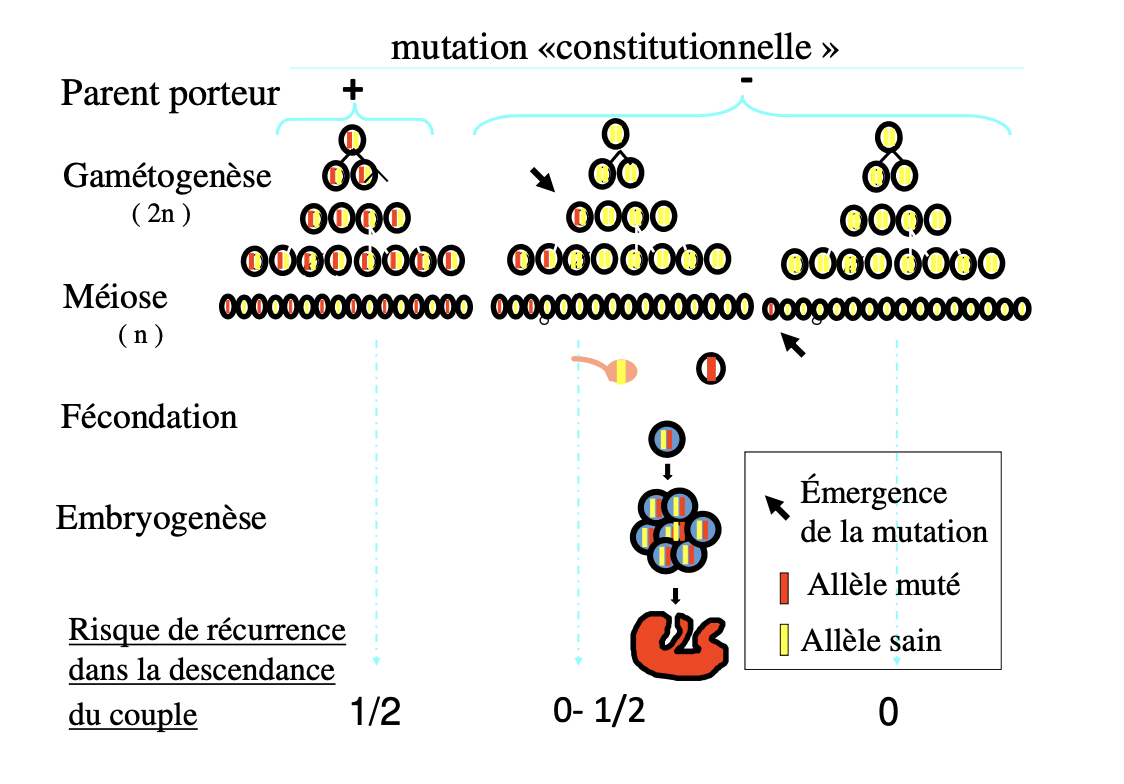
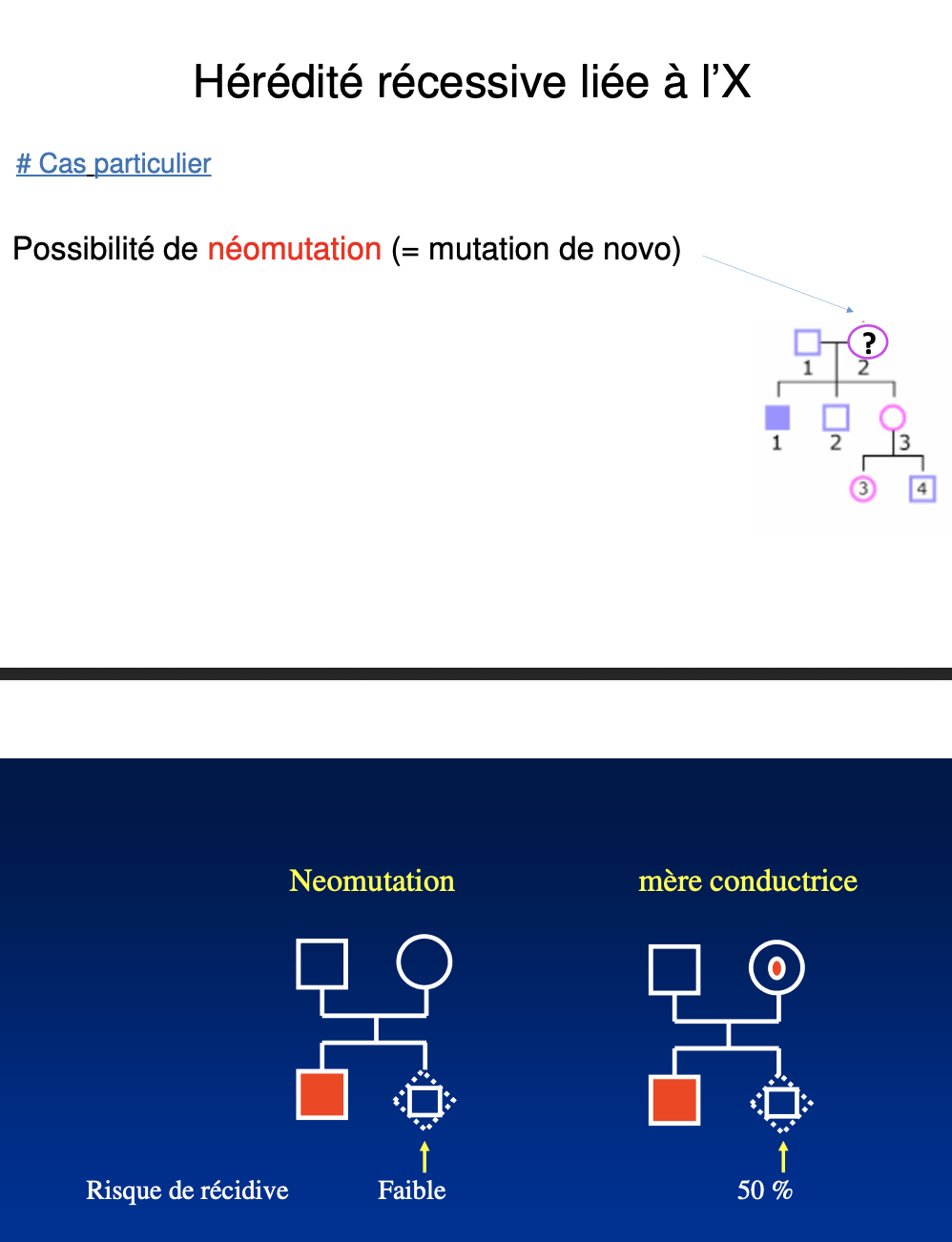
Qu’est-ce qu’une néomutation (mutation de novo) et quelles en sont les conséquences ?
Pourquoi dépister les femmes conductrices et par quels moyens cela peut-il se faire ?

L’hérédité récessive liée à l’X est une transmission où les caractères pathologiques sont portés par le chromosome X. Elle est dite récessive car la maladie ne s’exprime que si aucun allèle fonctionnel n’est présent, ce qui concerne surtout les garçons hémizygotes (un seul X). Les femmes hétérozygotes sont conductrices mais en général non malades, sauf en cas d’inactivation biaisée de l’X.
Si la mère est conductrice et le père sain :
50 % de risque d’avoir un garçon malade (X muté/Y).
50 % de risque d’avoir une fille conductrice (X muté/X sain).
Exemple : la myopathie de Duchenne.
Si le père est atteint et la mère saine :
Tous les garçons sont sains (car ils héritent du Y paternel).
Toutes les filles sont conductrices (X muté/X sain).
→ Cela entraîne une transmission "sautant une génération", avec risque pour les petits-fils.
La néomutation (mutation de novo) est une mutation apparaissant dans un gamète maternel. Elle peut provoquer un cas isolé chez un garçon atteint alors que les parents sont indemnes. Le risque de récurrence est très faible (~0), sauf en cas de mosaïcisme germinal.
Le dépistage des conductrices est essentiel pour évaluer le risque de récidive :
Phénotypage : recherche de signes cliniques mineurs (ex. augmentation de la CPK chez les conductrices de myopathie de Duchenne).
Génotypage : identification de la mutation chez le cas index puis recherche de cette mutation chez la mère.
Si la mutation est absente : néomutation → risque de récidive ~0 (sauf mosaïcisme germinal).
Si présente : risque de récidive de 50 % pour un fœtus de sexe masculin.
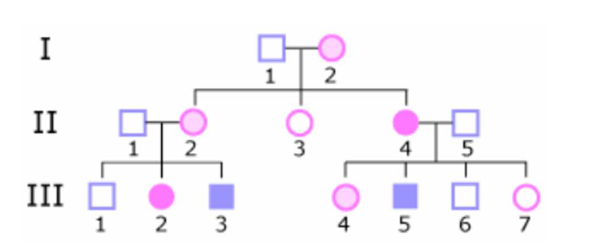
Risque pour II2 d’être conductrice? Ou Néomutation chez I1 ou I2?
déterminer si L’ X muté de II1 est d’origine paternelle ou maternelle
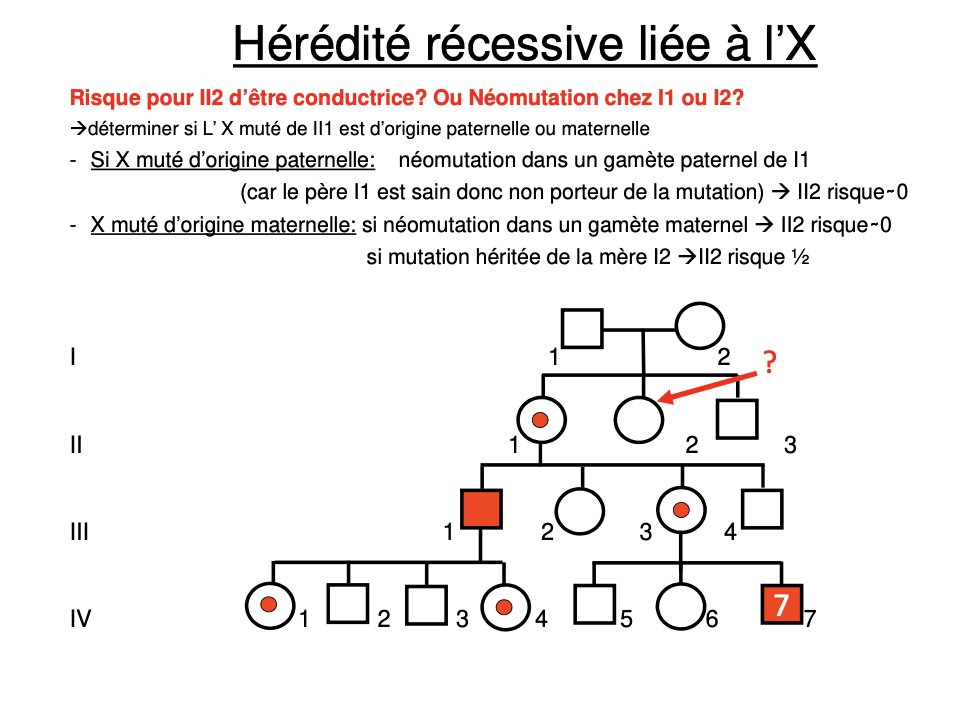
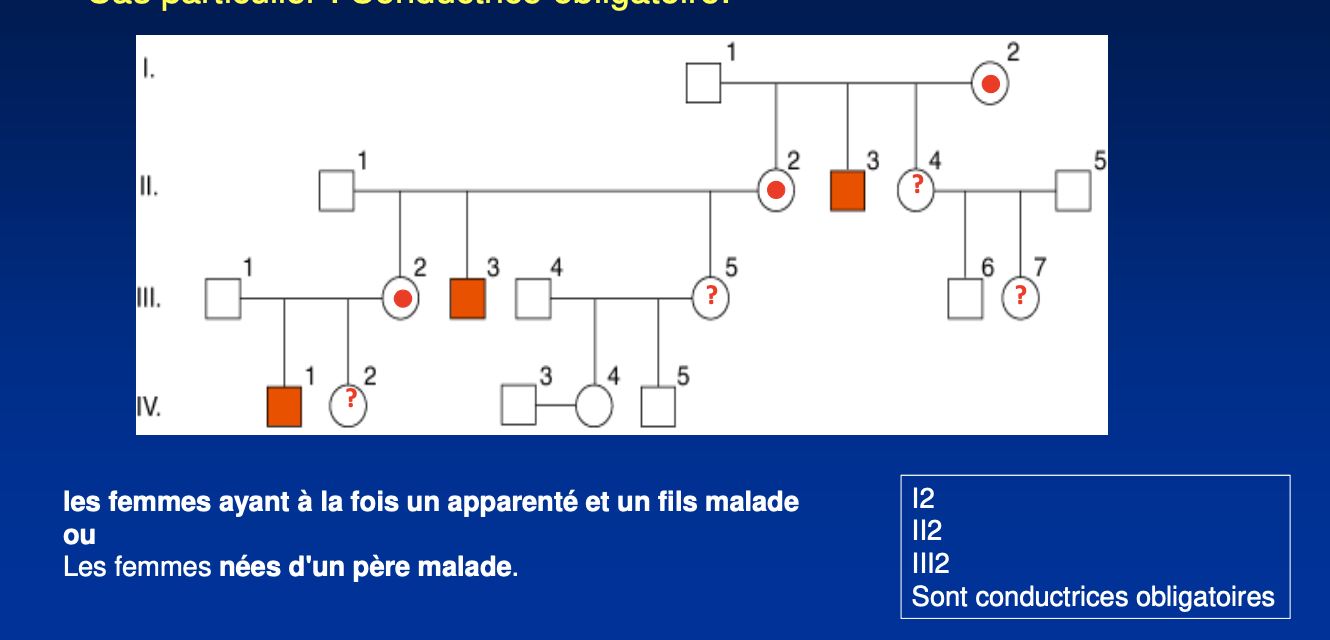
Qu’est-ce qu’un piège fréquent dans l’hérédité récessive liée à l’X en conseil génétique ?
Pourquoi certains hommes peuvent-ils être porteurs sains mais transmetteurs de la maladie ?
Comment une femme conductrice peut-elle devenir symptomatique ?
Quel est le lien entre inactivation de l’X et expression clinique de la maladie chez les femmes conductrices ?
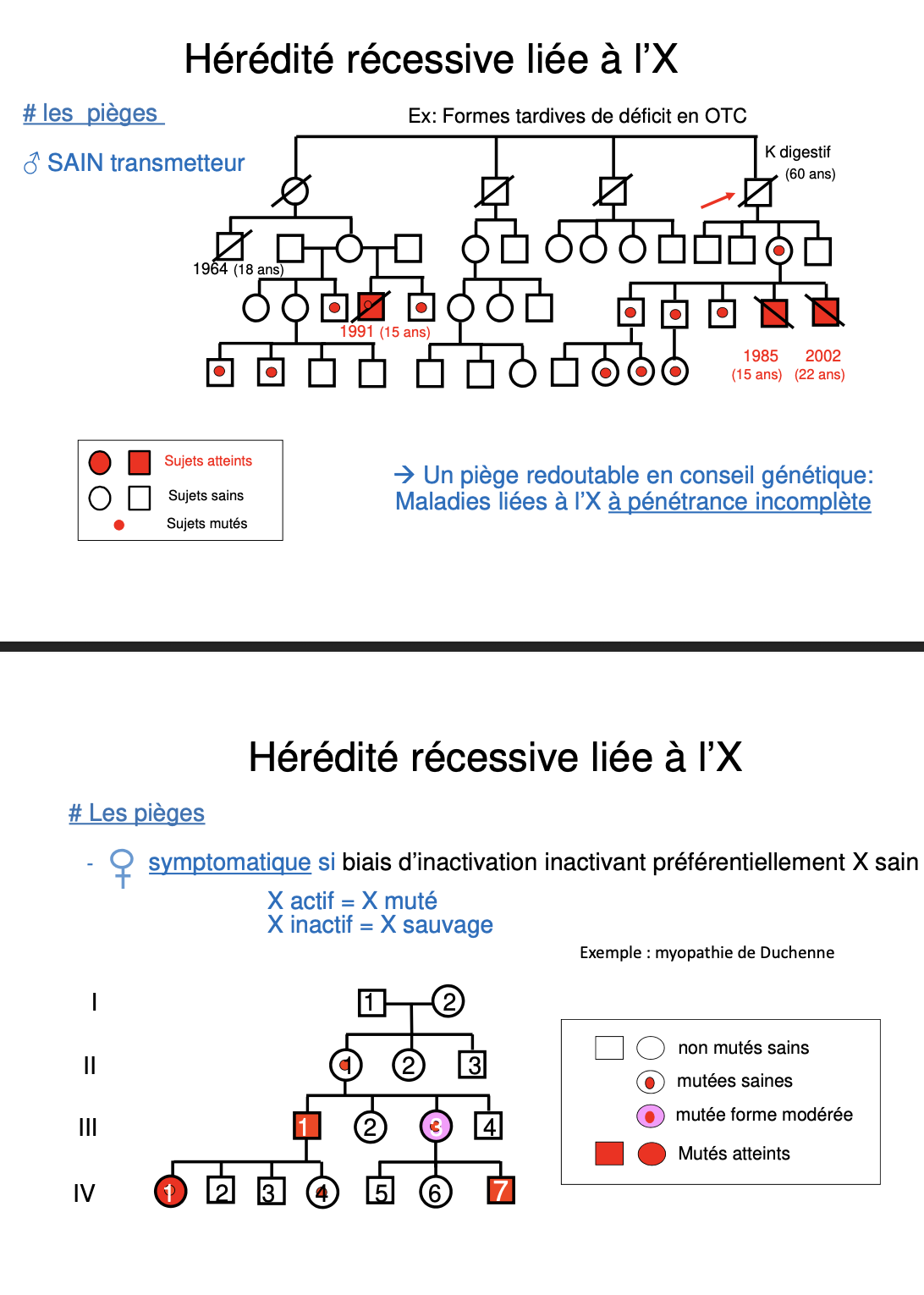
Un piège majeur est lié aux formes à pénétrance incomplète : certaines maladies RLX ne s’expriment pas systématiquement malgré la mutation, compliquant le conseil génétique et le dépistage dans les familles. Exemple : déficit tardif en OTC.
Certains hommes peuvent être sains transmetteurs lorsqu’ils portent une mutation qui reste silencieuse jusqu’à un âge tardif (formes atténuées). Ils ne présentent pas de symptômes précoces mais transmettent l’allèle muté à leurs filles, qui deviennent conductrices.
Une femme conductrice peut être symptomatique si l’inactivation de l’X se fait de façon biaisée. Si l’X porteur de l’allèle sain est majoritairement inactivé, l’X muté devient préférentiellement exprimé, provoquant l’apparition de signes cliniques (exemple : myopathie de Duchenne).
L’inactivation de l’X explique pourquoi certaines femmes hétérozygotes pour une mutation RLX peuvent être cliniquement atteintes.
Si X actif = X muté et X inactif = X sain, la femme présentera des symptômes.
Si l’inactivation est équilibrée ou favorise l’X muté inactif, la femme restera asymptomatique ou très peu atteinte.
im occ
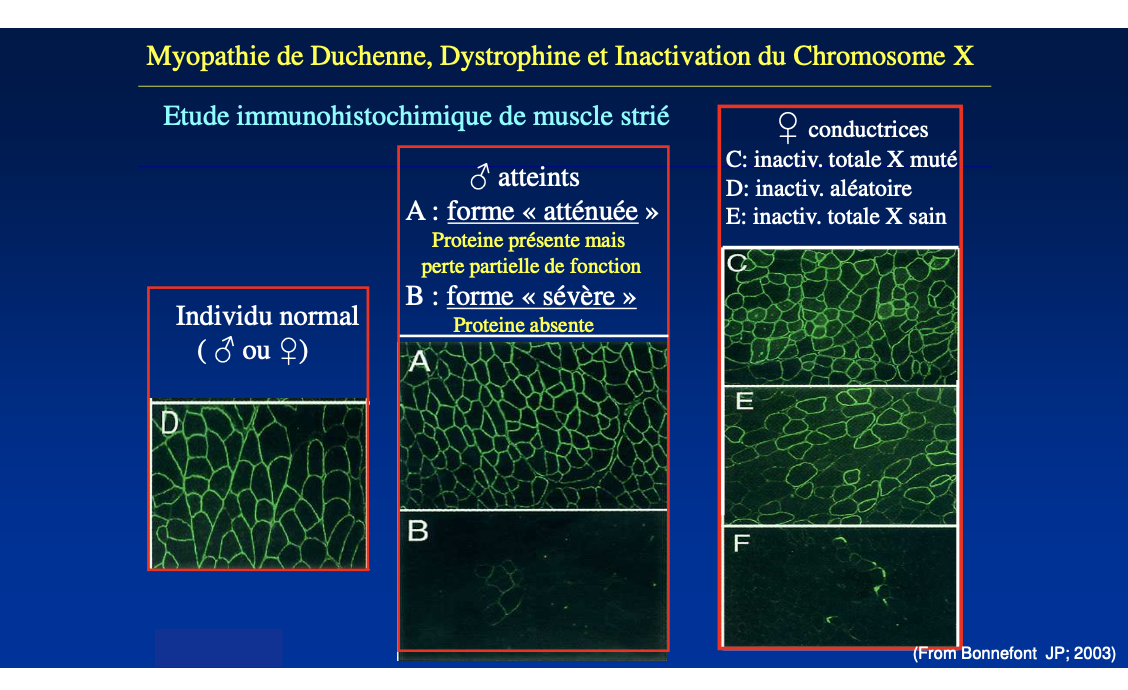
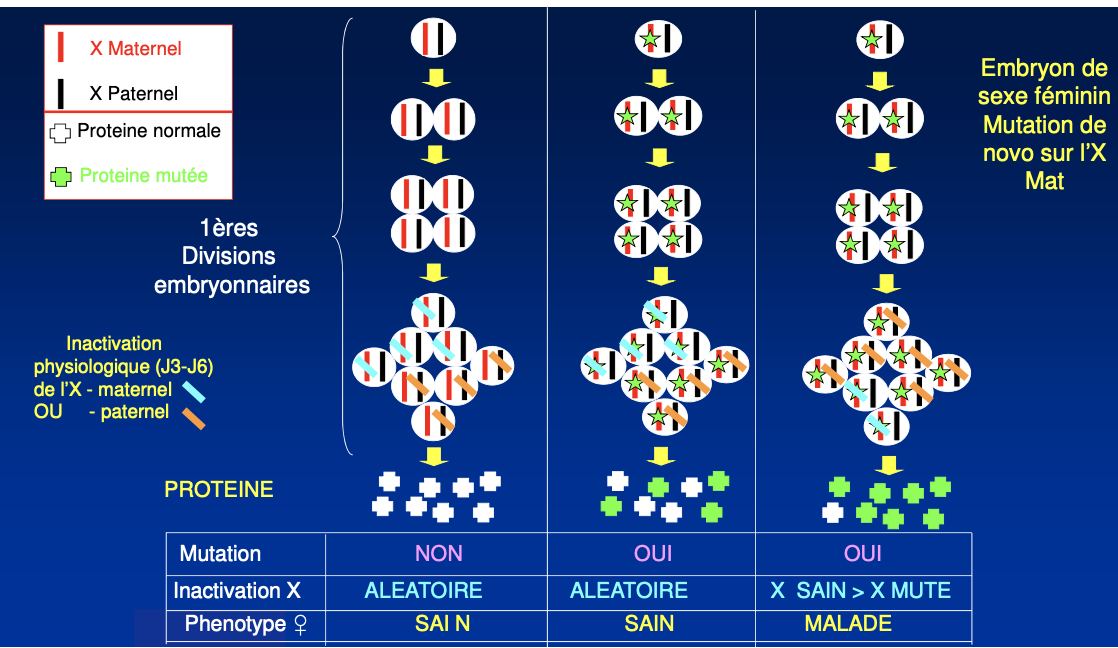
Quels sont les sexes le plus souvent atteints par une maladie récessive liée à l’X (RLX) ?
Comment la mutation responsable d’une RLX peut-elle être transmise ?
Quel est le risque de récurrence en cas de mutation de novo et quel rôle joue le mosaïcisme germinal ?
Existe-t-il une transmission père-fils dans les RLX ?
Comment peut-on dépister les femmes conductrices d’une RLX ?
Quels sont les risques de transmission dans la descendance d’une femme vectrice ?
Les femmes vectrices sont-elles toujours symptomatiques ?
Dans quelles conditions une femme vectrice peut-elle exprimer la maladie ?
Que se passe-t-il dans la descendance d’un homme transmetteur (porteur de la mutation sur son X) ?
Quel est le mécanisme moléculaire principal des maladies RLX ?
Qu’est-ce que la haploinsuffisance et quel rôle joue l’inactivation de l’X dans l’expression des RLX ?
Les sujets atteints de maladie RLX sont le plus souvent de sexe masculin, car ils sont hémizygotes pour le chromosome X et ne possèdent pas de copie saine de compensation.
La mutation peut être héritée de la mère conductrice ou bien apparaître de novo (néomutation) dans la lignée germinale.
En cas de mutation de novo, le risque de récurrence dans la fratrie est faible, mais non nul à cause d’un possible mosaïcisme germinal chez l’un des parents.
Il n’existe jamais de transmission père-fils dans les RLX, car un père transmet toujours son chromosome Y à ses fils.
Les conductrices peuvent être dépistées par phénotypage (ex. dosage enzymatique, signes cliniques minimes) ou par génotypage (mise en évidence directe de la mutation).
Dans la descendance d’une vectrice, il y a 1/2 de risque qu’un garçon soit atteint et 1/2 de risque qu’une fille soit conductrice.
Les femmes vectrices sont le plus souvent asymptomatiques, grâce à la présence d’un X sain qui compense.
Une femme vectrice peut être symptomatique si un biais d’inactivation de l’X désactive préférentiellement l’X sain, laissant s’exprimer l’X muté dans une majorité de cellules du tissu atteint.
Dans la descendance d’un homme transmetteur, tous les fils sont sains (puisqu’ils reçoivent le Y paternel) et toutes les filles sont conductrices (puisqu’elles reçoivent son X muté).
Les maladies RLX résultent généralement d’une perte de fonction d’une protéine (ex. dystrophine dans la myopathie de Duchenne).
La haploinsuffisance désigne la situation où un seul allèle fonctionnel ne suffit pas à assurer une activité normale. Chez les femmes, l’inactivation de l’X peut moduler le phénotype : si l’X sain est majoritairement inactivé, la maladie peut s’exprimer.
Hérédité non traditionnelle : Mosaïque Questions (retranscrites)
Mutation homogène vs mosaïque : définitions ?
Quand une mutation de novo donne-t-elle un phénotype homogène ou une mosaïque ?
Les formes de mosaïque possibles ?
Lien avec les gènes suppresseurs de tumeurs (principe des 2 événements) ?

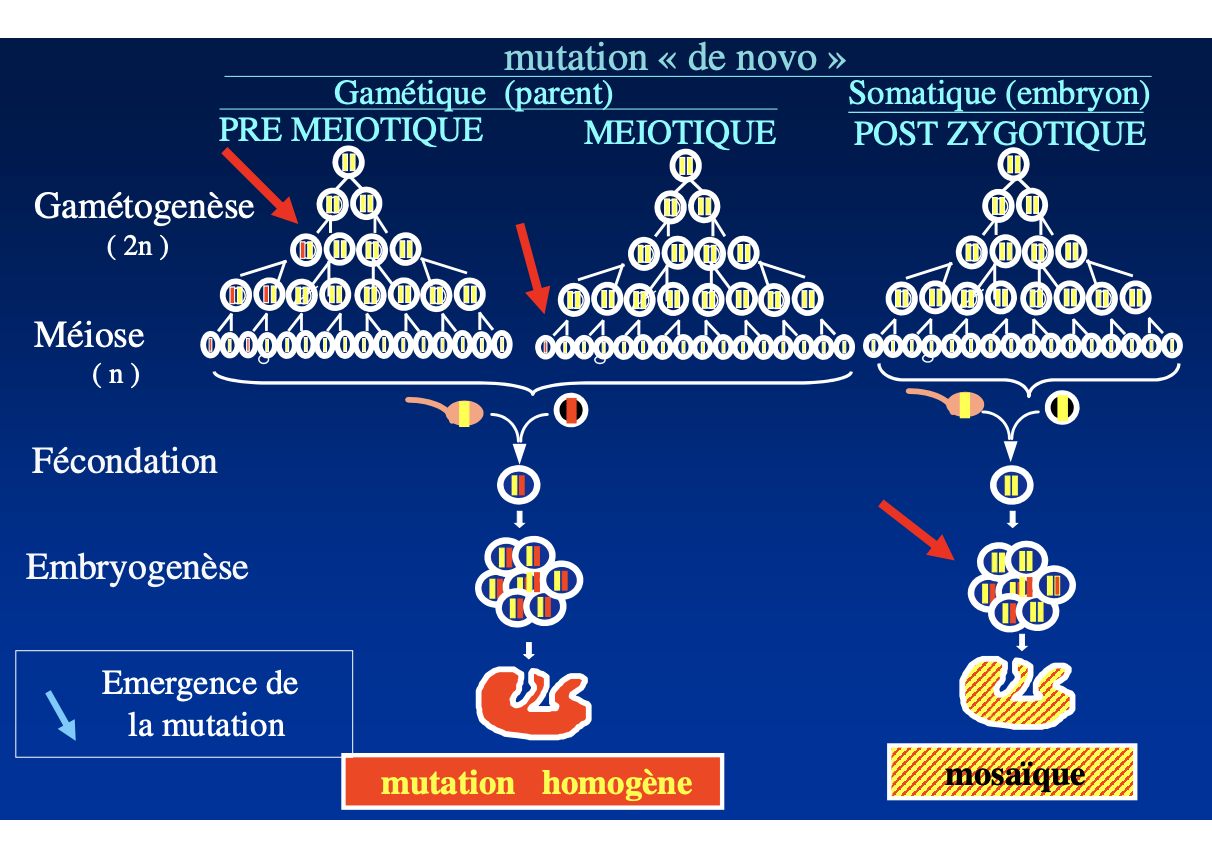
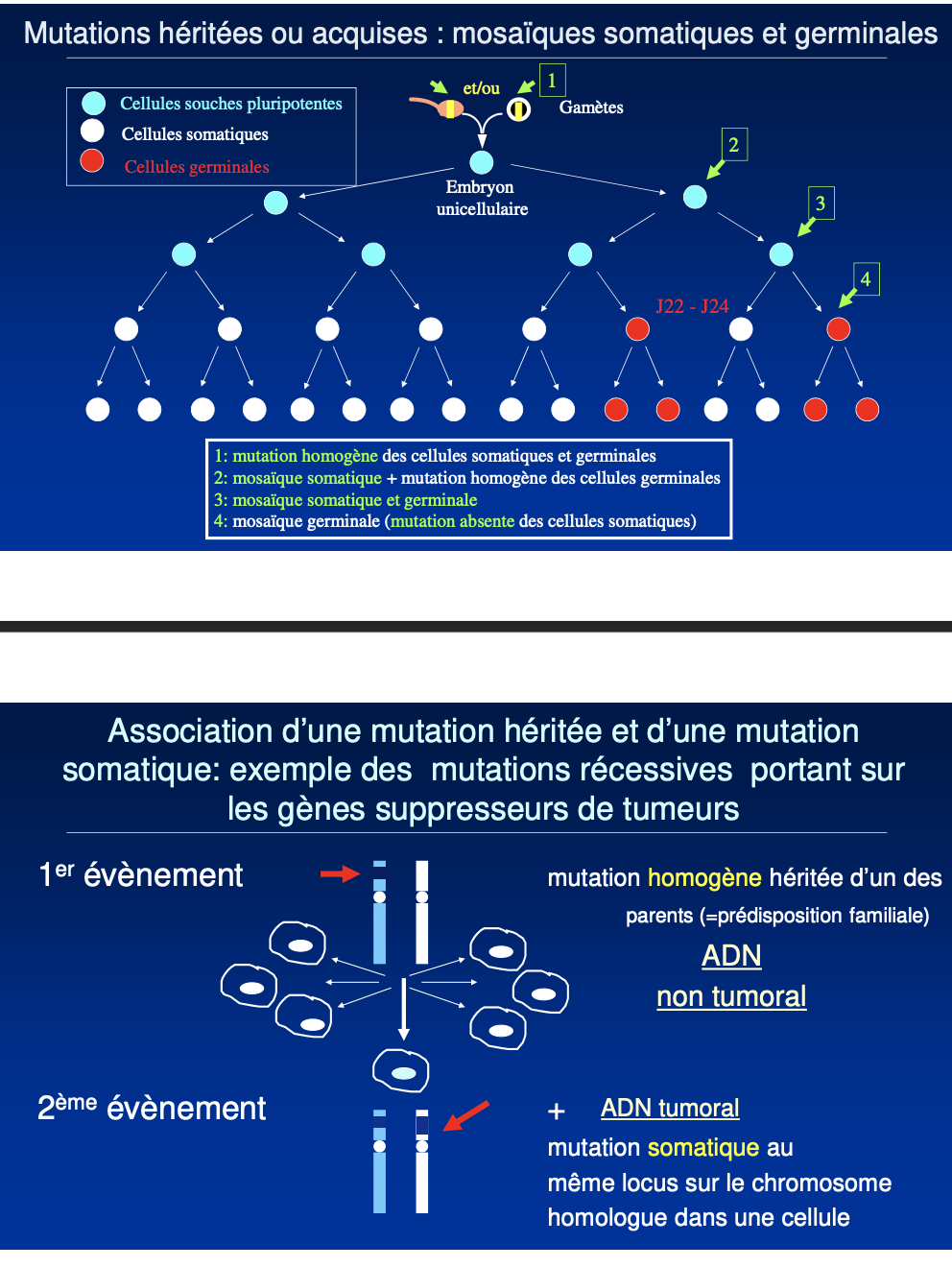
1) Définitions
Homogène : mutation dans toutes les cellules nucléées d’un individu.
Mosaïque : mutation présente seulement dans certaines cellules (certains tissus).
2) Origine de novo
Gamétique (pré-méiotique / méiotique, chez le parent) → gamète muté → embryon entier muté → mutation homogène.
Somatique post-zygotique (chez l’embryon) → mutation au cours des divisions embryonnaires → mosaïque.
3) Types de mosaïque
Somatique : limitée aux cellules somatiques (≠ germinales).
Germinale : limitée aux cellules germinales (absente des somatiques).
Somatique + germinale : les deux compartiments portent des clones mutés.
5) Gènes suppresseurs de tumeurs — 2 événements
1er événement (hérité) : individu hétérozygote pour une mutation germinale (ADN non tumoral).
2e événement (somatique) : perte/altération du deuxième allèle dans une cellule → cellule tumorale.
Hérédité non traditionnelle : Maladie à anticipation Questions (retranscrites)
Définition d’une maladie à anticipation ?
Mécanisme moléculaire commun ?
Exemples et motifs répétés associés ?
Fragilité de l’X (FMR1/FRAXA) : classes de répétitions, effets moléculaires ?
Risque de transition prémutation → mutation complète (ligne maternelle) ?


1) Définition
Maladie qui devient plus sévère et/ou se manifeste plus tôt au fil des générations (anticipation clinique).
2) Mécanisme commun
Expansion d’un segment d’ADN comportant des répétitions (souvent triplets, parfois quadruplets ou quintuplets).
Quand l’expansion dépasse un seuil, la maladie apparaît.
Peut concerner des gènes autosomiques ou liés à l’X.
3) Exemples (motif répété)
X fragile : (CGG)n
Maladie de Steinert (DM1) : (CTG)n
Ataxie de Friedreich : (GAA)n
Chorée de Huntington : (CAG)n
4) Fragilité de l’X — FMR1 (CGG)n
Allèle normal : < 50 CGG → mRNA +, protéine + → pas de symptômes.
Prémutation : 50–200 CGG → mRNA ++, protéine ± → symptômes ±.
Mutation complète : > 200 CGG → méthylation (CH₃) du gène, mRNA –, protéine – → FRAXA.
5) Risque de transition (prémutation maternelle → mutation complète dans la descendance)
55–59 : 0,5 %
60–70 : 10 %
70–80 : 31 %
80–90 : 70 %
90–100 : 80 %
101–200 : 98 %
🔑 Idée clé : plus la prémutation est grande, plus elle est instable → base moléculaire de l’anticipation.
Hérédité par expansion de triplets (MET) Partie A — Questions
Quels modes d’hérédité possibles ?
Qu’est-ce que l’instabilité intergénérationnelle ?
Quel phénomène clinique caractéristique apparaît au fil des générations ?
Les répétitions évoluent plutôt par expansion ou contraction ?
De quoi dépend l’amplitude du changement intergénérationnel chez un transmetteur ?
Quelle relation génotype–phénotype relie nombre de triplets et gravité ?
Ces maladies sont-elles en général de novo ou héritées ?
Quelle est la pénétrance habituelle ?
Qui peut être transmetteur sain et comment le détecter ?
Le mécanisme moléculaire relève d’une perte ou d’un gain de fonction ?

Autosomique dominante, récessive ou liée à l’X, selon la localisation du triplet.
Variation du nombre de répétitions (triplets/quadruplets…) d’une génération à l’autre.
Anticipation : sévérité accrue et/ou âge d’apparition plus précoce au fil des générations.
Majoritairement expansion (↑ du nombre), plus rarement contraction (↓).
Corrélée au nombre de triplets chez le sujet transmetteur (plus il est élevé, plus l’instabilité est grande).
Corrélation positive globale : plus de triplets → maladie plus sévère (avec seuils propres à chaque gène).
Plutôt héritées (les formes strictement de novo sont minoritaires).
Souvent incomplète (notamment pour plusieurs formes liées à l’X ou dominantes).
Des porteurs de pré-mutation cliniquement sains ; détection par génotypage uniquement.
Selon le gène : perte de fonction (silencing, haplo-insuffisance) ou gain de fonction toxique (ARN/protéine).
3/ hérédité non traditionnelle
Maladie par mutation d’un gène soumis à empreinte
Qu’est-ce que l’empreinte génomique ?
L’expression d’un gène soumis à empreinte est-elle bi- ou monoallélique ? De quoi dépend-elle ?
À quel moment l’empreinte s’installe, s’efface et se réappose ? Est-elle stable ?
Quelles sont les caractéristiques générales des gènes soumis à empreinte (proportion, organisation, tissus) ?
Quelles sont les bases moléculaires de l’empreinte (mécanismes épigénétiques majeurs) ?
Signes moléculaires d’un gène inactif vs actif (ADN/histones/chromatine) ?
Quel est le rôle des ARN non codants dans l’empreinte ?
Quelles règles d’hérédité « non mendélienne » découlent de l’empreinte (pénétrance, variabilité, parent transmetteur) ?
Pour un gène à expression maternelle exclusive, qui transmet le phénotype si porteur de la mutation ? Et quels risques pour la descendance ?
Même question pour un gène à expression paternelle exclusive.
Une mutation peut-elle être transmise par les deux parents ? La maladie dépend-elle de l’origine parentale ?
Donnez un exemple : causes possibles d’un syndrome d’Angelman (gène UBE3A).
Que signifie UPD paternelle dans ce contexte ?



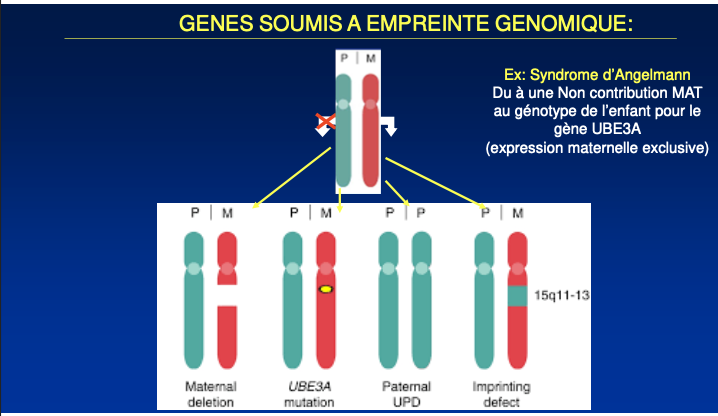
Phénomène par lequel l’expression d’un gène dépend de l’origine parentale (maternelle ou paternelle) du chromosome qui le porte.
Monoallélique : un seul allèle est exprimé, l’autre est inactivé selon l’origine parentale.
L’empreinte est posée pendant la gamétogenèse (spécifique du sexe), effacée chez l’enfant dans sa lignée germinale puis réapposée selon son sexe. Elle est stable au cours des mitoses.
Elle concerne une minorité de gènes, souvent regroupés en clusters, et peut être tissu-spécifique (ex. placenta, développement embryonnaire/fœtal).
Mécanismes épigénétiques : méthylation de l’ADN (îlots CpG/DMR-ICR), modifications des histones (acétylation/désacétylation, etc.) et remodelage de la chromatine; participation d’ARN non codants.
Gène inactif : ADN méthylé, histones désacétylées, chromatine condensée.
Gène actif : ADN déméthylé, histones acétylées, chromatine ouverte.
Des ARN non codants (imprinted lncRNA, etc.) participent à l’installation/au maintien de l’empreinte et à la répression d’un des allèles.
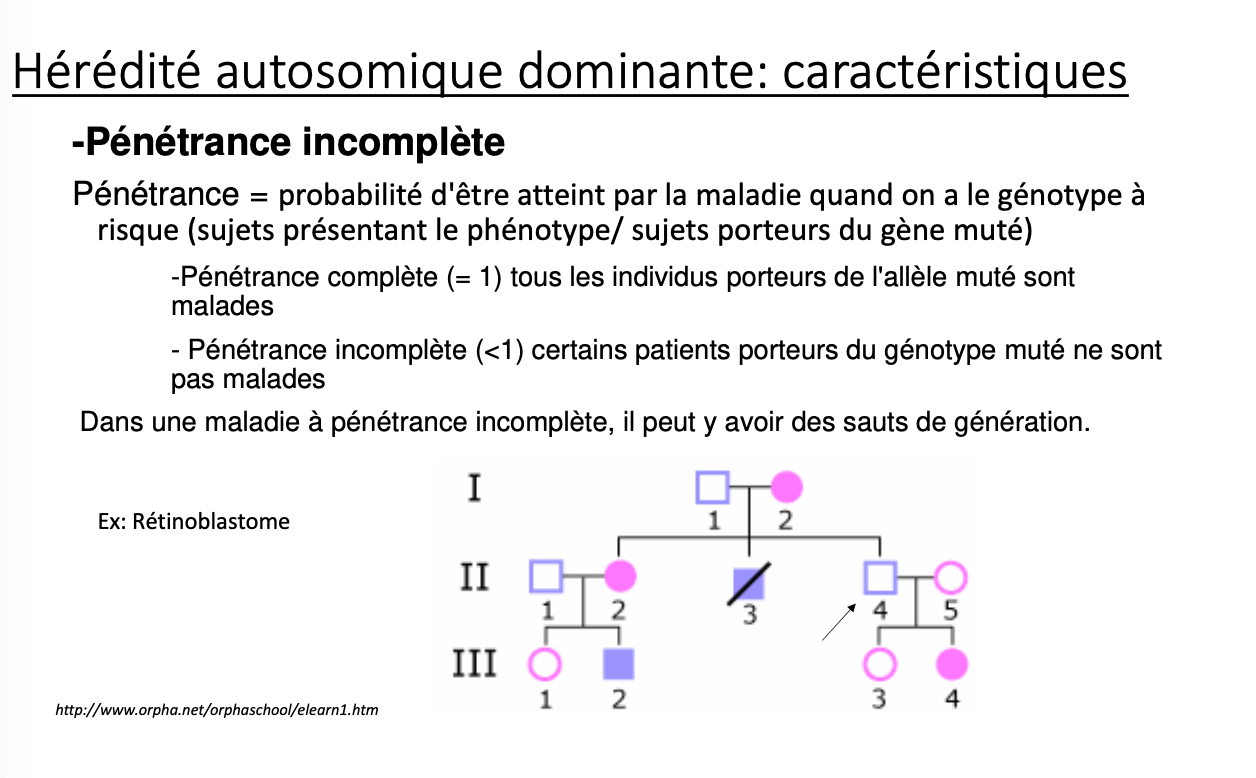
Hérédité non mendélienne : pénétrance incomplète, variabilité d’expressivité, et surtout dépendance au sexe du parent transmetteur. On observe des sauts de génération apparents lorsque la mutation est transmise par le parent dont l’allèle est normalement silencé.
Expression maternelle exclusive (allèle paternel silencé) :
Mère mutée : transmet la mutation et le phénotype (≈ 1/2 enfants atteints).
Père muté : transmet la mutation sans phénotype chez ses enfants (allèle paternel silencé).
Expression paternelle exclusive (allèle maternel silencé) :
Père muté : transmet la mutation et le phénotype (≈ 1/2 enfants atteints).
Mère mutée : transmet la mutation sans phénotype (allèle maternel silencé).
Oui, la mutation peut être transmise par l’un ou l’autre parent, mais la maladie ne s’exprime que si la mutation se trouve sur l’allèle normalement exprimé (dépend de l’origine parentale).
Syndrome d’Angelman (UBE3A, expression maternelle dans le cerveau) : perte de contribution maternelle par
délétion maternelle de 15q11-q13,
mutation de l’allèle maternel UBE3A,
UPD paternelle (deux chromosomes paternels 15),
défaut d’empreinte (ICR).
UPD paternelle = Uniparental Disomy d’origine paternelle : l’enfant reçoit deux copies paternelles du chromosome 15 (et aucune maternelle) → absence d’expression maternelle de UBE3A → phénotype Angelman.
L’empreinte génomique est-elle pathologique ou physiologique ?
En quoi consiste l’empreinte génomique au niveau de l’expression des allèles ?
L’empreinte concerne-t-elle tout le génome ? Si non, quoi précisément ?
L’empreinte peut-elle être tissu-spécifique ?
Quand l’empreinte s’efface et se réappose-t-elle ? De quoi cela dépend-il ?
Est-elle stable au cours des mitoses somatiques ?
Quels mécanismes moléculaires assurent l’empreinte ?
D’où peut venir une mutation d’un gène soumis à empreinte ?
Pourquoi le phénotype dépend-il de l’origine parentale du chromosome porteur ?
Citez les grands types d’anomalies responsables de maladies des gènes soumis à empreinte.

Physiologique.
Expression monoallélique d’un gène en double copie, déterminée par l’origine parentale (pat ou mat) du chromosome.
Non : elle touche certaines régions de certains chromosomes (ex. 7, 11, 14, 15…).
Oui, l’empreinte peut être tissu-spécifique.
Elle est effacée puis réapposée d’une génération à l’autre dans l’ADN des cellules germinales, pendant le développement intra-utérin, selon le sexe fœtal.
Oui, l’empreinte est stable au cours des mitoses des cellules somatiques.
Mécanismes épigénétiques : méthylation de l’ADN, modifications des histones (acétylation/désacétylation…), rôle d’ARN non codants.
Soit héritée d’un parent transmetteur, soit de novo.
Parce qu’un seul allèle est actif : si la mutation touche l’allèle normalement exprimé (selon qu’il est d’origine maternelle ou paternelle), la maladie s’exprime ; sinon elle peut rester silencieuse.
Macro-délétions, disomies uniparentales (UPD), mutations géniques des gènes soumis à empreinte, altérations du centre de contrôle de l’empreinte (ICR).
3/ hérédité non traditionnelle
Maladie par mutation de l’ADN mitochondrial
Quelle est la règle d’hérédité des mutations de l’ADN mitochondrial (ADNmt) ?
Qui peut être atteint ? Qui peut transmettre ?
Qu’est-ce que l’hétéroplasmie ? À quoi est-elle corrélée ?
Qu’appelle-t-on homoplasmie ?
Pourquoi parle-t-on de « double origine génétique » des mitochondries ?
Combien de produits code l’ADNmt et lesquels (grandes catégories) ?
D’où viennent les autres sous-unités de la chaîne respiratoire mitochondriale ?
Quel gamète apporte les mitochondries à l’embryon ?
Chez l’homme porteur d’une mutation de l’ADNmt, la descendance est-elle à risque ?
Comment se manifeste la transmission familiale sur un arbre généalogique ?
Que signifient « expressivité variable » et « pénétrance incomplète » pour ces maladies ?
Le risque et la sévérité de la maladie varient avec quel paramètre clé ?
Donner un principe simple pour reconnaître une transmission mitochondriale dans une famille.
Pourquoi des femmes cliniquement saines peuvent-elles avoir des enfants atteints ?
Quel est l’impact de la ségrégation mitotique de l’ADNmt au cours des divisions cellulaires ?


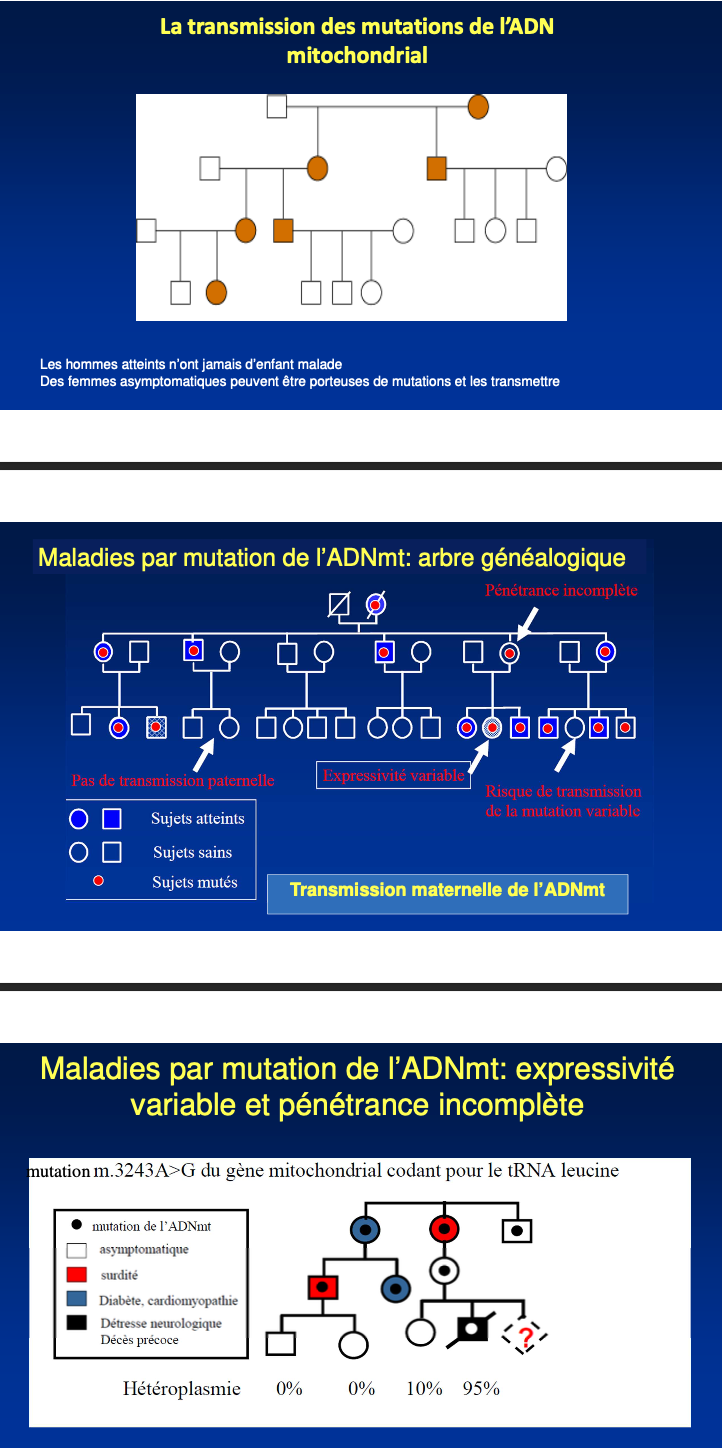
Hérédité exclusivement maternelle.
Hommes et femmes peuvent être atteints, seules les femmes (mères) transmettent.
Hétéroplasmie = coexistence, dans une même cellule/tissu, d’ADNmt sauvage et muté ; son taux est corrélé au risque et à la sévérité de la maladie.
Homoplasmie = 100 % d’ADNmt d’un même type (tout sauvage ou tout muté).
Les mitochondries comportent des sous-unités codées par le génome nucléaire et par l’ADNmt.
L’ADNmt code 13 polypeptides de la chaîne respiratoire + 22 tRNA et 2 rRNA.
Les autres sous-unités (≈ 70 polypeptides) sont codées par l’ADN nucléaire.
Seul l’ovocyte maternel apporte les mitochondries (et l’ADNmt) à l’embryon.
Non. Les hommes atteints/porteurs ne transmettent jamais la mutation.
Tous les enfants d’une mère porteuse sont à risque, alors qu’aucun enfant d’un père atteint ne l’est ; la transmission suit la ligne maternelle.
Expressivité variable : intensité des symptômes différente selon les sujets ; pénétrance incomplète : certains porteurs ne présentent aucun symptôme.
Le taux d’hétéroplasmie (proportion d’ADNmt muté) dans les tissus concernés.
Recherche d’une absence de transmission paternelle et d’une agrégation par la lignée maternelle (mères, oncles maternels, cousins maternels…).
À cause de l’hétéroplasmie et de la pénétrance incomplète : elles peuvent être porteuses sans symptômes mais transmettre des charges mutées plus élevées.
La ségrégation mitotique peut modifier la proportion d’ADNmt muté dans les cellules filles, expliquant les variations tissulaires et familiales du phénotype.
Maladies par mutation de l’ADN mitochondrial
Quelle est la particularité de l’hérédité des mutations d’ADNmt ?
Qui peut être atteint et qui transmet ?
Qu’appelle-t-on hétéroplasmie ?
Qu’est-ce que la ségrégation mitotique de l’ADNmt ?
Comment le taux d’hétéroplasmie influence-t-il la maladie ?
Quel enchaînement moléculaire survient quand le seuil d’hétéroplasmie est dépassé ?
Quels indices généalogiques font penser à une transmission mitochondriale ? (3 idées)
En quoi une atteinte mitochondriale peut-elle mimer une autosomique dominante, et quel point la contredit ?
En quoi peut-elle mimer une récessive liée à l’X, et quel point la contredit ?
En quoi peut-elle mimer une dominante liée à l’X, et quel point la contredit ?
Quelle est la double origine génétique de la chaîne respiratoire mitochondriale (éléments codés par chaque génome) ?
Quelles caractéristiques cliniques de population observe-t-on souvent pour ces maladies (2 idées) ?


Transmission exclusivement maternelle des mitochondries.
Hommes et femmes peuvent être atteints ; seules les femmes transmettent. Les hommes atteints ne transmettent jamais.
Coexistence de plusieurs populations d’ADNmt (sauvage et muté) dans une mitochondrie, une cellule ou un tissu.
À partir d’une cellule hétéroplasme, apparition de clones où un seul type d’ADNmt peut prédominer après divisions.
Le risque et la sévérité augmentent avec le taux d’hétéroplasmie ; expression si un seuil tissulaire est dépassé.
Déficit de la chaîne respiratoire → ↓ production d’ATP + accumulation de radicaux libres toxiques → souffrance tissulaire.
a) Absence de transmission paternelle ; b) toute la descendance d’une mère peut être à risque ; c) expressivité variable / pénétrance incomplète.
Mimique autosomique dominante (atteints sur plusieurs générations) mais : jamais de transmission par les pères.
Mimique récessive liée à l’X mais : sex-ratio des atteints ≈ 1:1.
Mimique dominante liée à l’X mais : sévérité indépendante du sexe.
ADN nucléaire : ~70 polypeptides des complexes ; ADN mitochondrial : 13 polypeptides + 22 tRNA et 2 rRNA.
Expressivité variable et pénétrance incomplète ; risque de transmission variable selon le niveau d’hétéroplasmie.

Quel génome possèdent les mitochondries ?
Quelle est la taille et la nature de l’ADNmt, et combien de gènes contient-il ?
Que codent les gènes mitochondriaux (détail par catégories) ?
En combien de complexes est organisée la chaîne respiratoire et quelle est l’origine génétique de leurs sous-unités ?
Combien de copies d’ADNmt trouve-t-on par cellule ?
Comment s’appelle la coexistence de plusieurs populations d’ADNmt chez un individu ?
Comment s’appelle la présence d’une seule population d’ADNmt ?
Quel est le mode de transmission intergénérationnelle des mitochondries ?
De quoi dépendent la pénétrance et l’expressivité des maladies mitochondriales ?
Quels modes d’hérédité les maladies mitochondriales peuvent-elles mimer ?
