PHSI3010 Module 2: Cardiovascular System (Final Exam)
1/60
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
61 Terms
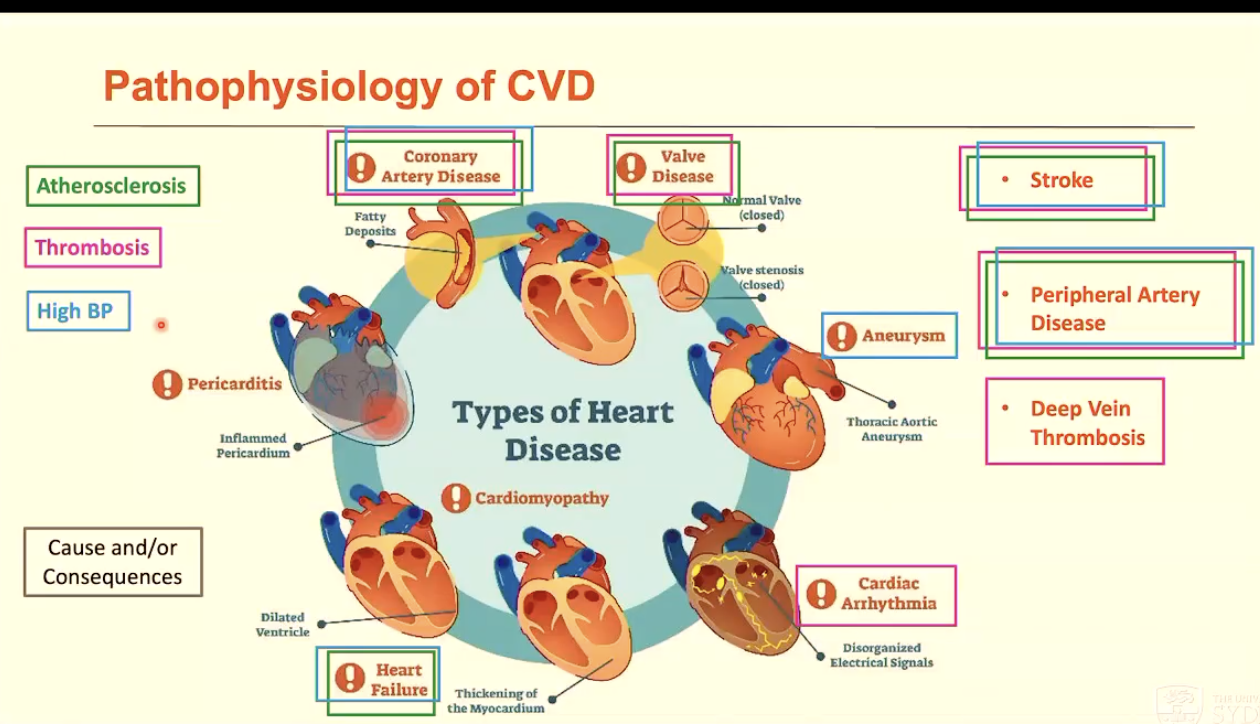
List a range of cardiovascular diseases (10)
Coronary/Ischemic heart disease: disease of coronary vessels
Stroke: disease of blood vessels supplying brain
Peripheral vascular disease: disease of blood vessels supplying limbs
Heart failure and cardiomyopathy: disease of heart muscle
Aortic aneurysm: enlargement, weakness, and rupture of aortic wall
Venous thrombosis: blood clots in the leg veins (deep vein thrombosis), which can dislodge and move to heart and lungs (pulmonary embolism)
Valve disease: leaky or narrow valves
Hypertensive disease: high BP
Rheumatic heart disease: damage to heart muscle and valves from rheumatic fever (caused by streptococcal bacteria)
Congenital heart disease: birth defects that affect normal development and functioning of heart
How many deaths does CVD account for?
17.9 million deaths / year worldwide (~32% of all deaths)
highest: heart attack (second: stroke)
What are the main risk factors of CVD?
Non-modifiable risk factors
age, sex, genetics
Modifiable risk factors (if you have >2, you are at risk of CVD)
smoking
alcohol
unhealthy diet
physical inactivity
diabetes
obesity
high cholesterol
high BP
Why is CVD such a problem (3)?
Increasing incidence of diabetes and obesity
Up to 25% of people who have CAD don’t have risk factors
Improved survival from MI means more people are living with damaged myocardium and implants
What are the underlying pathologies of CVD (3)?
Atherosclerosis
Thrombosis
High BP
How is ECM involved in CVDs?
ECM remodelling/dysregulation is a critical step in a number of CVDs, leading to:
hypertension and arterial stiffness (contributes to CVD)
aortic aneurysm
heart failure
What is hypertension?
high BP > 140/90
What are the 2 types of hypertension?
Primary hypertension = 90% of cases (unknown cause)
normal CVD risk factors (e.g. hereditary diet, obesity, smoking, alcohol, stress, smoking, diabetes)
causes unknown but involves one or a combination of:
genetics
nervous system/baroreceptor
stress (leads to a higher risk of a heart attack in the morning)
salt overconsumption (increased water retention → increased CO and BP)
obesity (increased insulin resistance → more salt sensitive)
can be reduced: cumulative effect of reducing multiple factors
Secondary hypertension (known cause)
Caused by renal and adrenal diseases, obstructive sleep apnea, preeclampsia, pulmonary hypertension (no need to know these specific diseases)
What are the consequences of hypertension?
Hypertension leads to end-organ damage (e.g. kidney, heart, brain)
but can also lead to earlier damage: endothelial damage and arterial stiffening
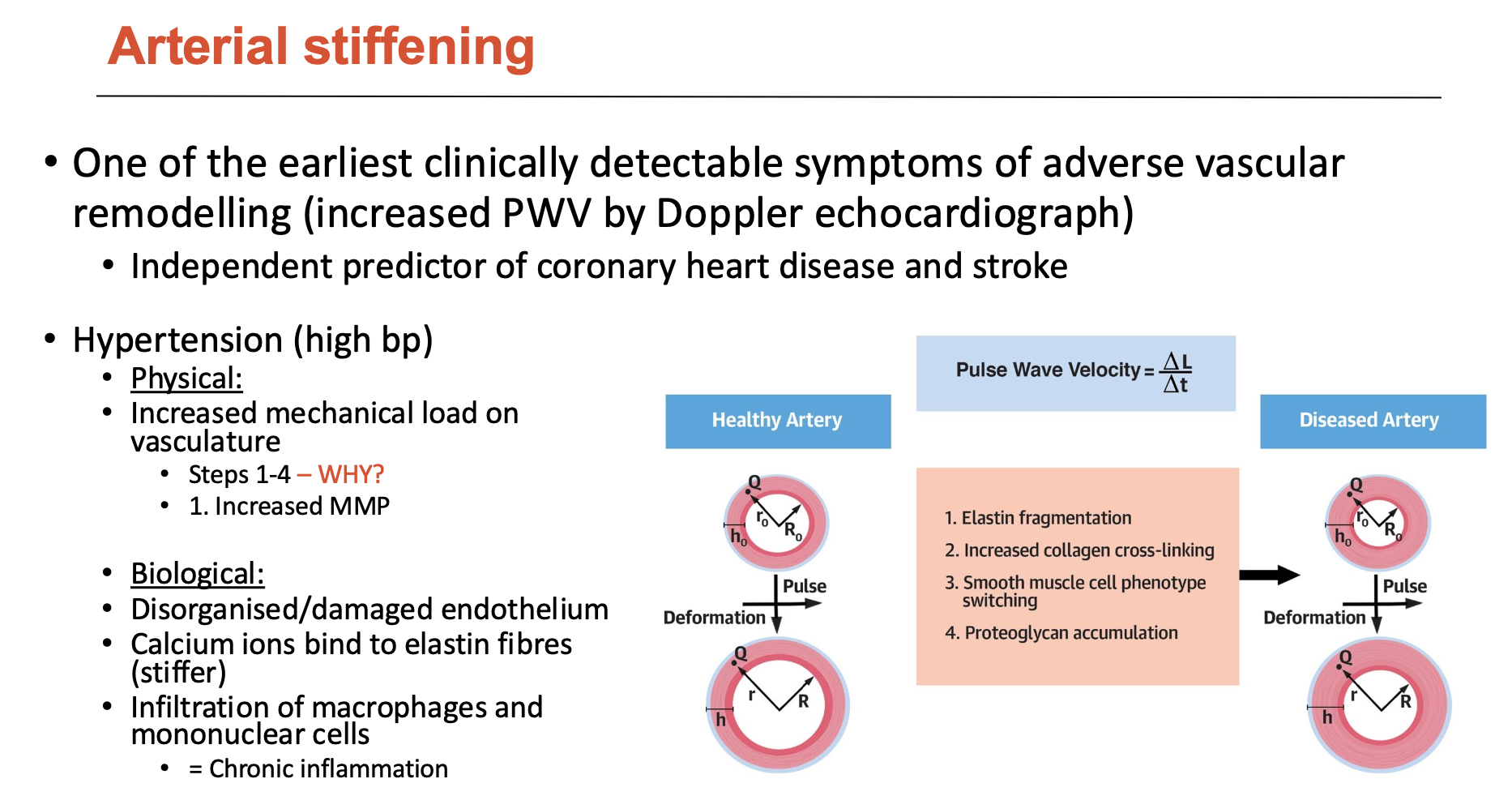
What does arterial stiffening indicate?
Arterial stiffening is an independent predictor of coronary heart disease and stroke; leads to high BP, which causes:
increased mechanical load on vasculature (physically)
disorganised/damaged endothelium (biologically)
calcium ions bind to elastin fibres (stiffer)
infiltration of macrophages ad mononuclear cells = chronic inflammation
This stimulates:
elastin fragmentation
increased collagen cross-linking
SMC phenotype switching
proteoglycan accumulation
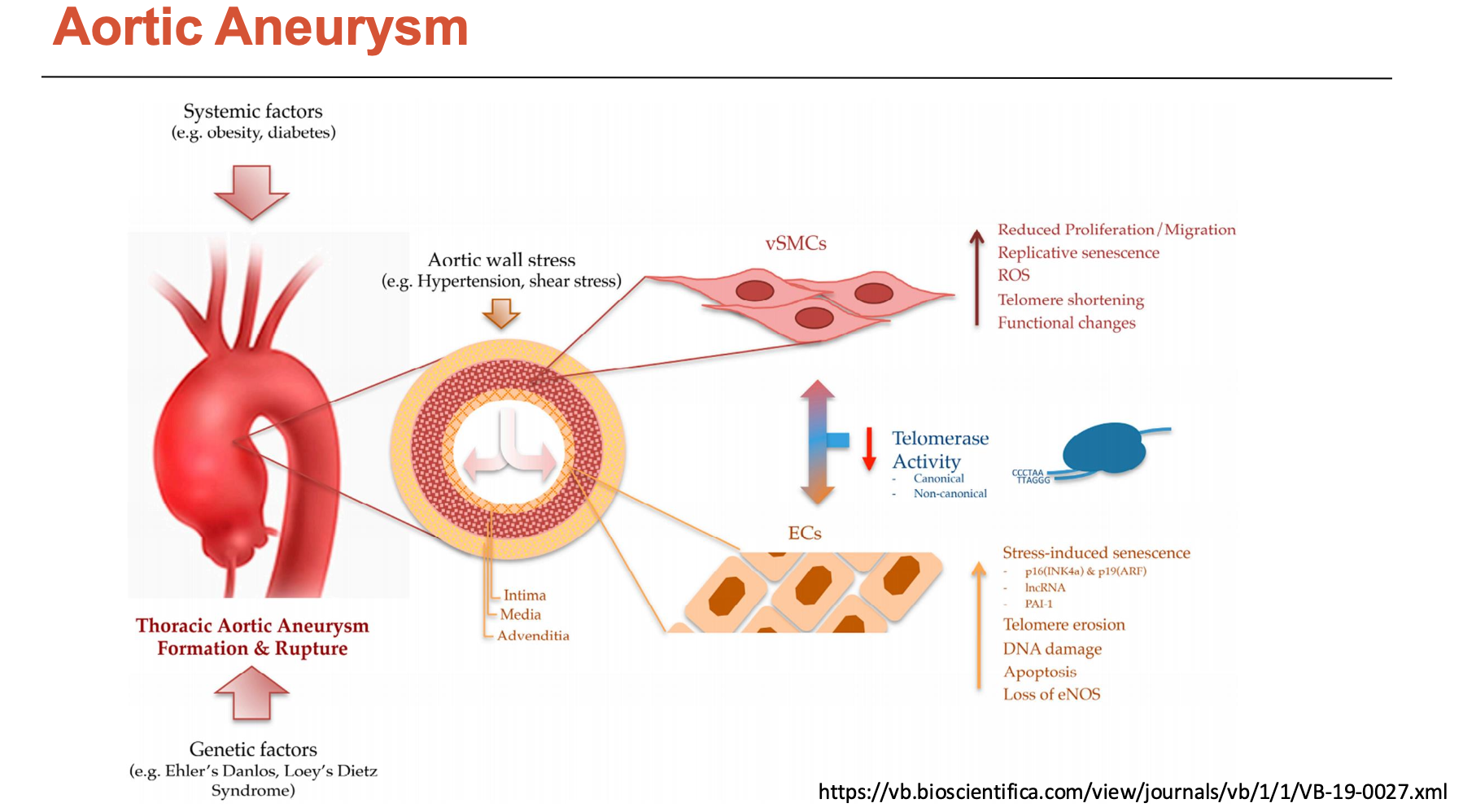
What happens in an aortic aneurysm?
There are two types of aortic aneurysms:
Thoracic aortic aneurysm: above diaphragm (less prevalent)
Abdominal aortic aneurysm: below diaphragm (more prevalent)
Its pathophysiology involves high BP consequences at a quicker rate:
medial degeneration
SMC loss (apoptosis) and contractile to synthetic phenotype switch
increased MMP production
cytokine release (e.g. TGF-beta)
elastic fibre fragmentation
disorganised collagen
accumulated proteoglycans
What are the 5 stages of atherosclerosis?
Endothelial dysfunction
Lipid accumulation
Recruitment of leukocytes into the vessel wall, foam cell formation
Fibrous plaque
Plaque rupture
Note: the first 3 can occur simultaneously
What are the 6 steps of atherosclerosis?
Endothelial cells become activated, attracting blood leucocytes (monocytes, dendritic cells, T-cells) to adhere to the activated endothelial monolayer
Bound leucocytes migrate into the intima
When a monocyte enters the intima, it matures into a macrophage, then transforms into a foam cell upon consuming lipids (foam cells localise fatty deposits on blood vessels - more likely in diabetics who cannot uptake fats)
foam cells release retention molecules, cytokines, and chemokines, which further progress inflammation
As the lesion progresses, more smooth muscle cells migrate from media into intima to form a bump (plaque structure)
Apoptotic plaque macrophages and SMCs have extracellular lipids that can accumulate in the central region of the plaque (necrotic lipid core)
Note: VSMCs can also become macrophage-like foam cells → contribute to necrotic core
Plaque becomes unstable (thinning of fibrous cap) → vessel ruptures → platelet aggregation/thrombosis formation → can impede blood flow OR obstruct another organ’s blood flow (infarction → stroke)
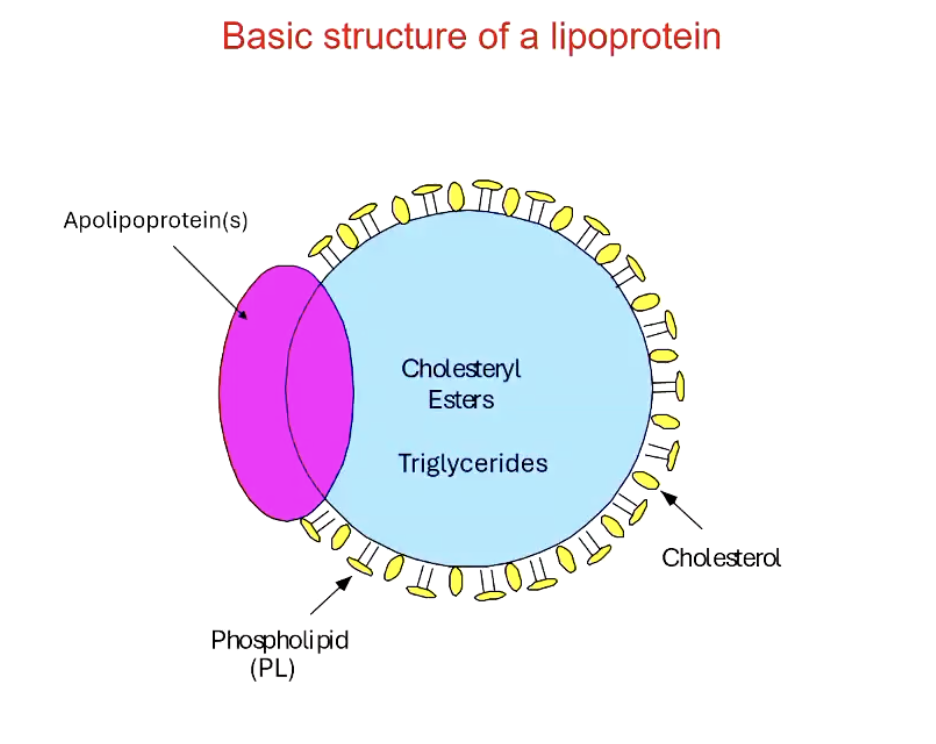
What is the basic structure of a lipoprotein?
Spherical, core has neutral lipids like cholesteryl esters and TGs
surrounded by phospholipids and free cholesterol
associated with apolipoproteins that direct lipoproteins to target tissues
What are the functions of lipoproteins?
Serve to transport lipid-soluble compounds b/w tissues
substrates for energy metabolism (TGs)
essential components for cell growth and division (phopholipids, cholesterol)
precursors for hormones (cholesterol)
precursors for eicosanoids (FAs)
lipid-soluble vitamins (vitamin E, beta-carotene)
precursors for bile acids (cholesterol)
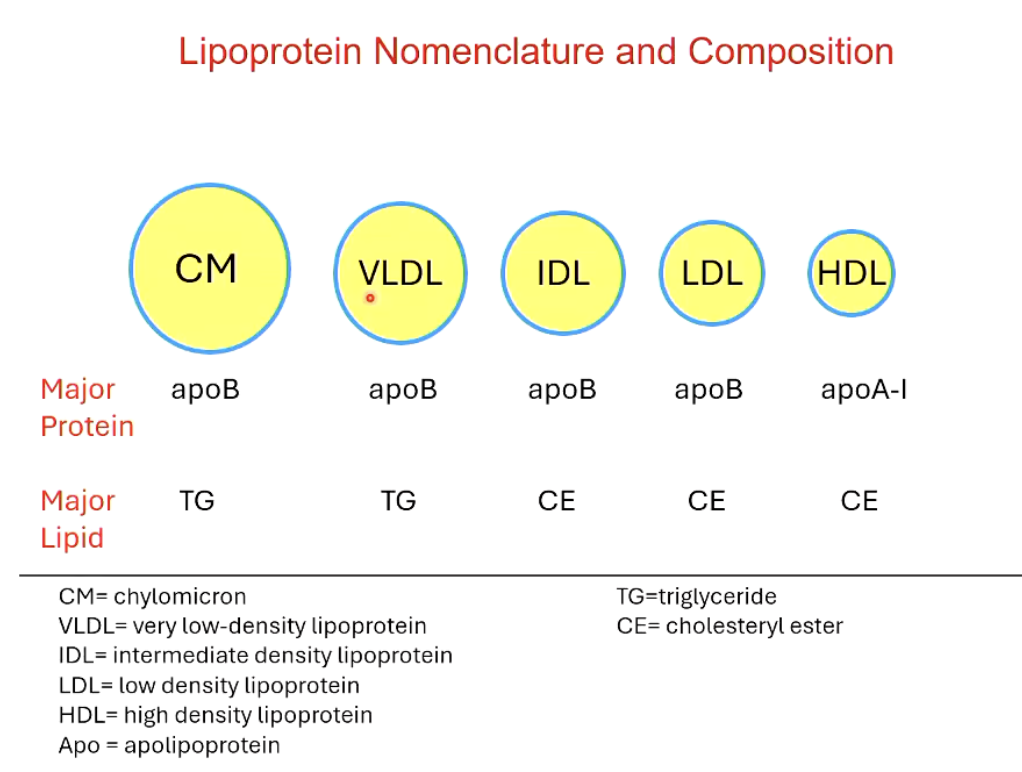
What are the 5 different classes of lipoproteins?
CM (chylomicron): apoB & TG; intestinal origin
VLDL (very low density lipoprotein): apoB & TG; liver origin
IDL (intermediate density lipoprotein): apoB & CE; liver origin
LDL (low density lipoprotein): apoB & CE; liver origin
HDL (high density lipoprotein): apoB & CE; liver and intestinal origin
What are the actions of lipoproteins in vivo?
Liver secretes VLDL in the bloodstream → IDL → LDL by losing TGs to peripheral tissues
LDL is taken up by the liver
CMs are secreted by the intestines and release FAs into peripheral tissues, leaving chylomicron remnants (CMR) left → is taken up by liver
Intestine and liver also secrete HDL (good cholesterol) to collect free cholesterol from peripheral tissue and take it up into the liver, to break it down
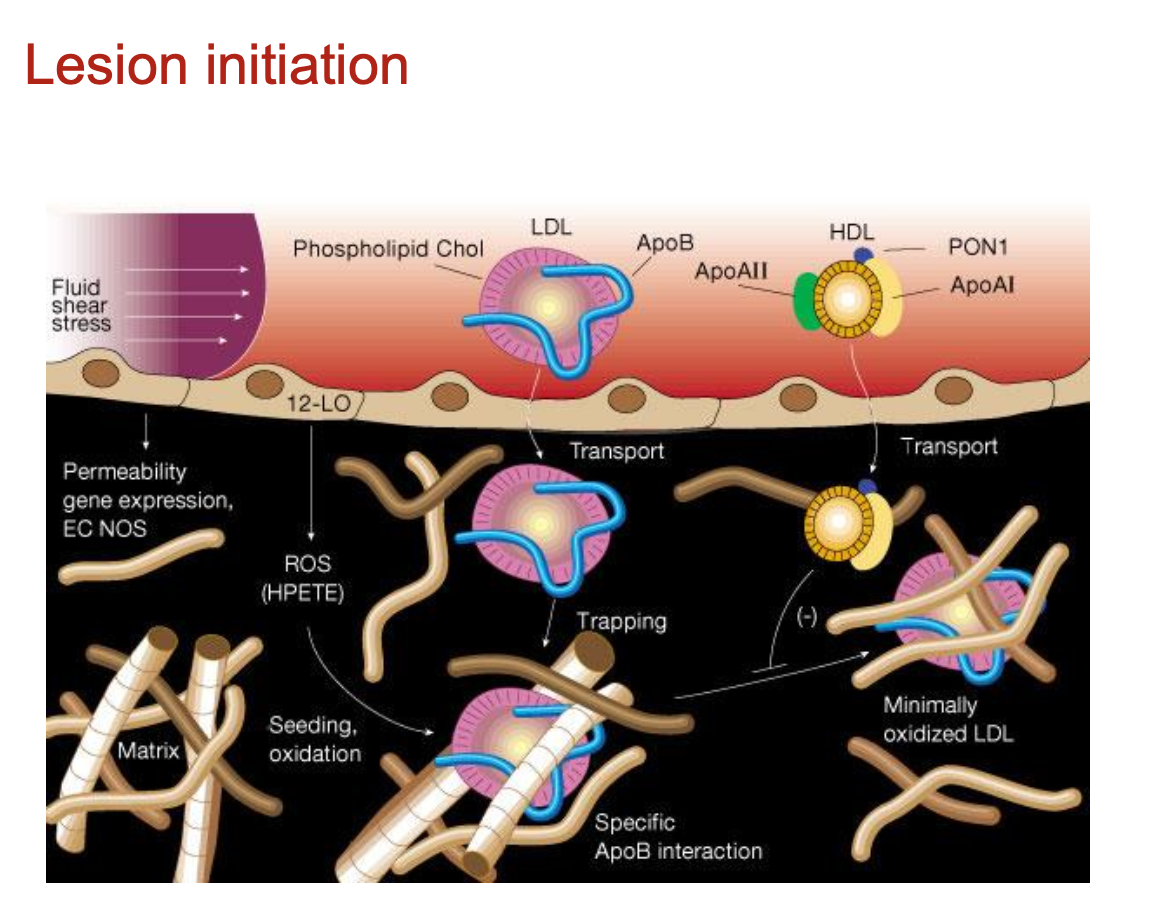
How are atherosclerotic lesions initiated?
Atherosclerosis begins with dysfunctional endothelium and retention of ApoB100-containing lipoproteins (VLDL, IDL, LDL) into the sub-intimal space
several other apolipoproteins (e.g. apoE) have proteoglycan-binding domains and may also be retained in the vessel wall
What is the ApoE-/- mouse model?
ApoE is a glycoprotein that serves as a ligand for receptors that clear cholesterol from the blood; maintains cholesterol homeostasis
is also involved in immune regulation (e.g. it is produced by monocyte/macrophages)
ApoE-/- mice have impaired cholesterol metabolism and subsequently, increased plasma cholesterol
however, their lipid profile is non-human like
ApoE-/- mice develop atherosclerosis spontaneously, which is accelerated on a high fat “Western”-style diet
How do macrophages become foam cells?
Through scavenger receptors, macropinocytosis, or phagocytosis of aggregated LDL
foam cells have impaired lipid efflux
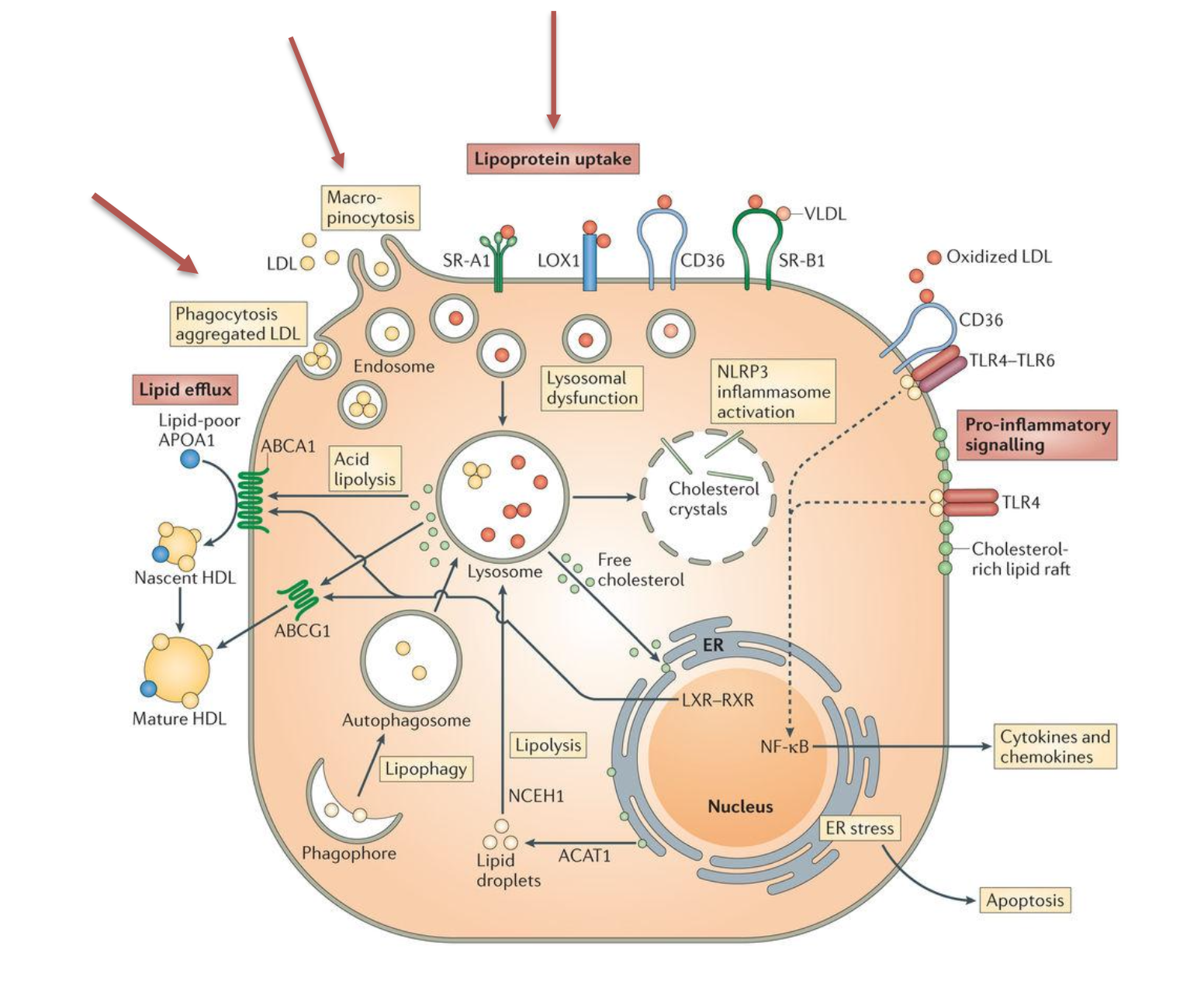
How does elevated cholesterol affect ATP-binding cassette family (ABCs)?
Accumulation of cholesterol activates the liver X receptor (LXR)-retinoid X receptor(RXR) heterodimeric TF, which upregulates ABCA1, ABCG1
these mediate transfer of free cholesterol to lipid poor ApoA1 to form nascent/mature HDL
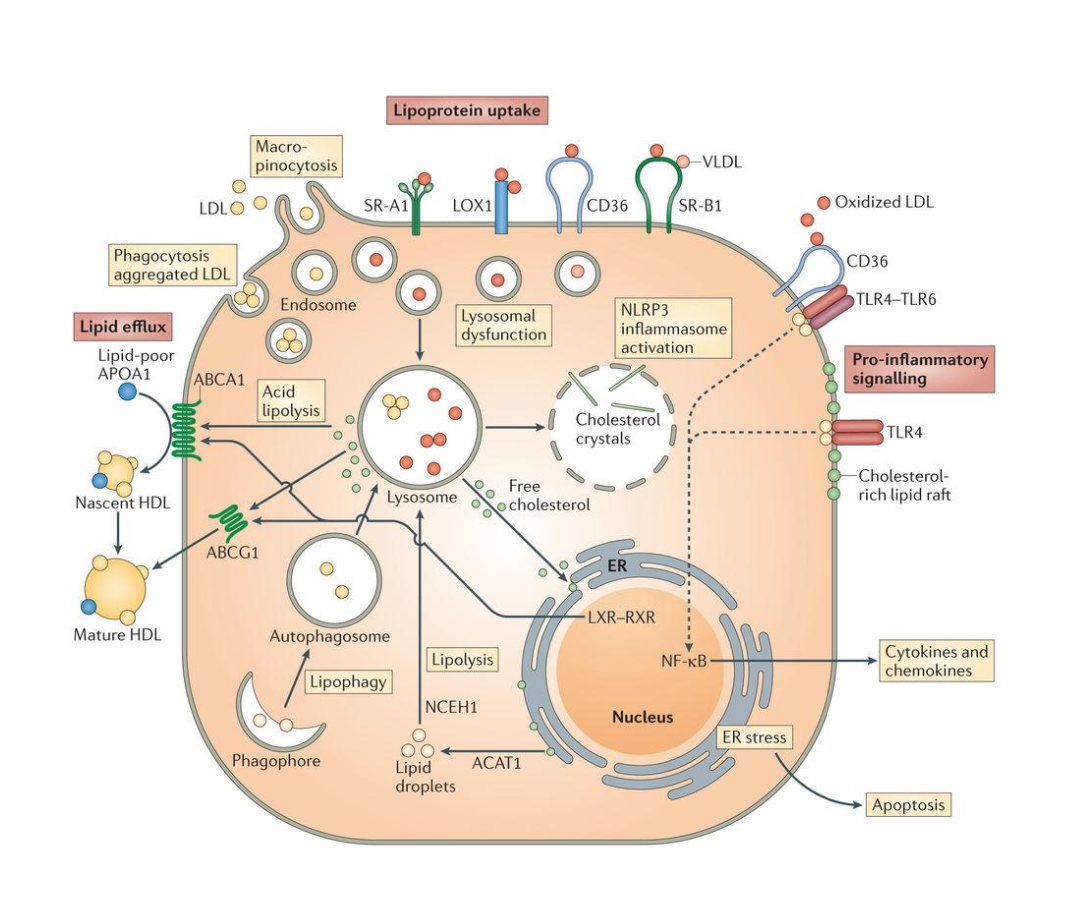
What are the consequences of defective cholesterol trafficking in macrophages?
Inflammation: accumulation of cholesterol in cell membranes enhances inflammatory signalling (e.g. TLRs and activation of NF)
Cell death: apoptosis, autophagy, and necroptosis
Defective efferocytosis: the process by which dead/dying cells are cleared by macrophages
What are the mechanisms of plaque rupture?
Plaque rupture occurs where the cap is thinnest and most infiltrated by foam cells. Thinning of the fibrous cap can occur via 2 ways:
gradual loss of SMCs from the fibrous cap (usually bc of cell death)
infiltrating macrophages degrade the collagen-rich cap matrix
These mechanisms can occur simultaneously
What are the mechanisms of statins?
Statins inhibit the conversion of acetate → cholesterol, by inhibiting the rate-limiting enzyme: HMG-CoA reductase
lowers cell cholesterol levels → induces synthesis of LDL receptors
plasma LDL is taken up by LDL receptors
LDL uptake by cells lowers plasma LDL levels (promotes LDL clearance)
How does colchicine work against atherosclerosis?
Colchicine inhibits microtubule polymerisation → arrests the spindle action during mitosis
What are some recent drugs that are potential therapies for atherosclerosis?
Canakinumab leads to residual inflammatory risk and increased risk of sepsis
Not such a great idea to target one cytokine critical for innate immunity?
Colchicine is the first approved drug targeting inflammation by inhibiting microtubule polymerisation → arrests the spindle action during mitosis
Long history of clinical use, minimal adverse effects, inexpensive
but doesn’t work for everyone
What are the 3 steps of hemostasis?
Hemostasis occurs to clot the bleeding of a blood vessel
Vasoconstriction
Platelet plug
Clot reinforcement = coagulation (fibrin formation)
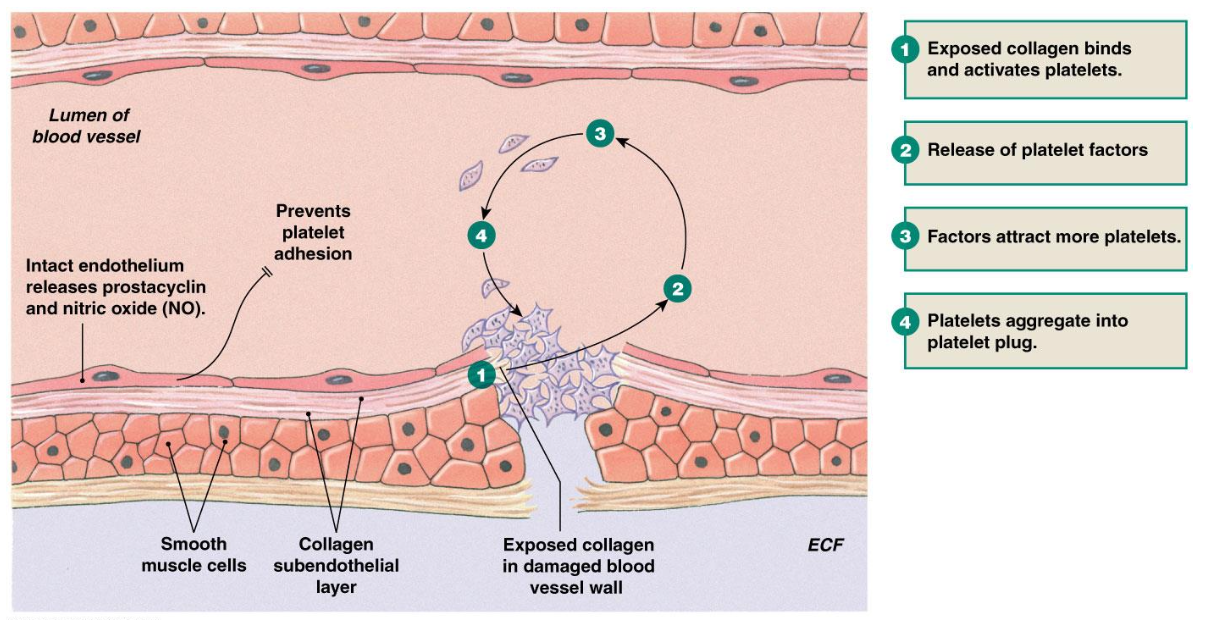
How are platelet plugs formed in hemostasis (4)?
Exposed collagen binds and activates platelets
Release of platelet factors
Factors attract more platelets
Platelets aggregate into platelet plug
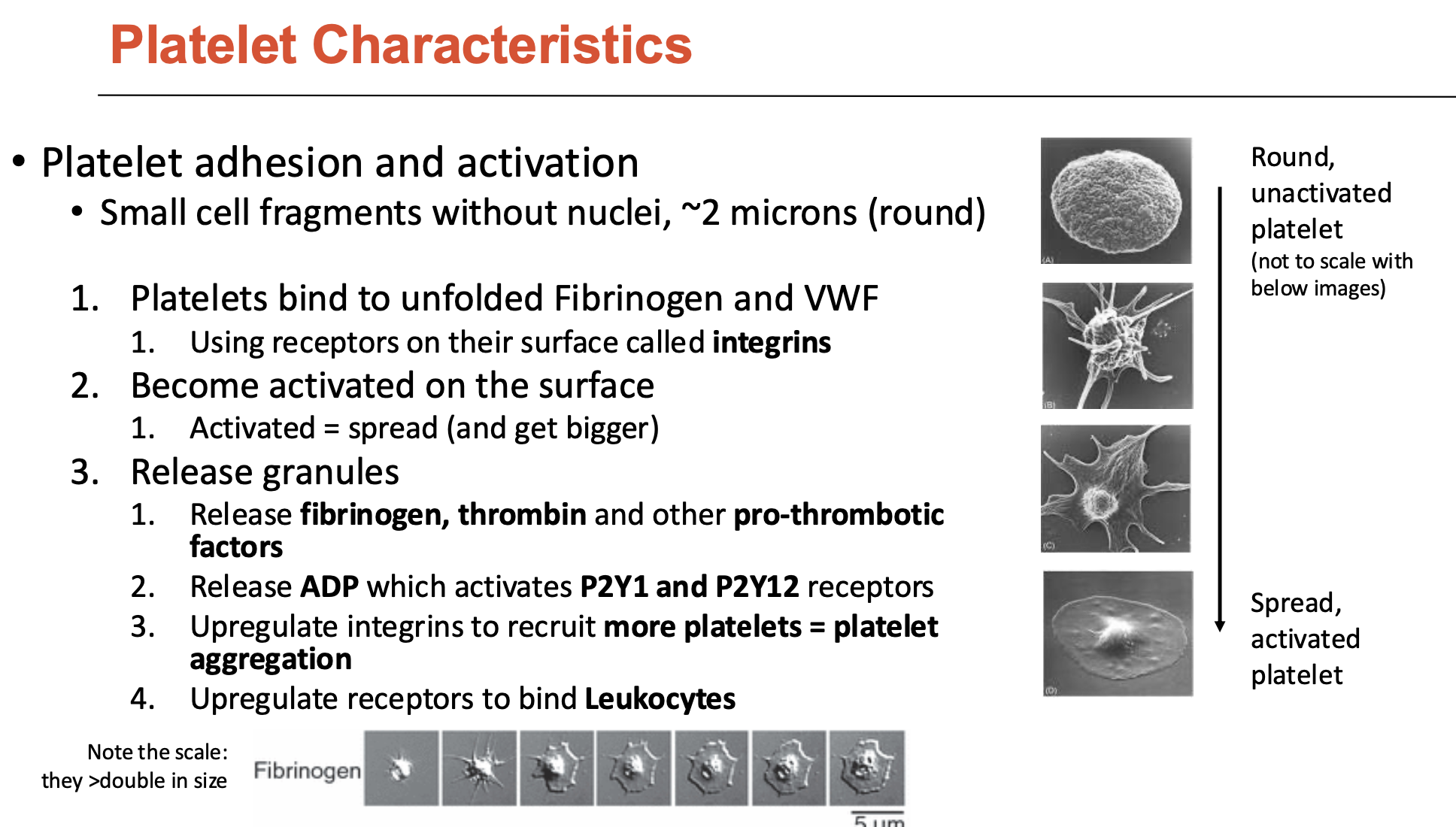
What are the characteristics of platelets?
Platelets are small cell fragments without nuclei (~2 microns)
have thousands of receptors on their surface
How do platelets undergo adhesion and activation (3)?
Platelets bind to unfolded fibrinogen and vWF
uses surface receptors called integrins
Become activated on the surface
activated = spread (and get bigger)
Release granules
release fibrinogen, thrombin, and other pro-thrombotic factors
release ADP → activates P2Y1 and P2Y12 receptors
upregulate integrins to recruit more platelets = platelet aggregation
upregulate receptors to bind leukocytes
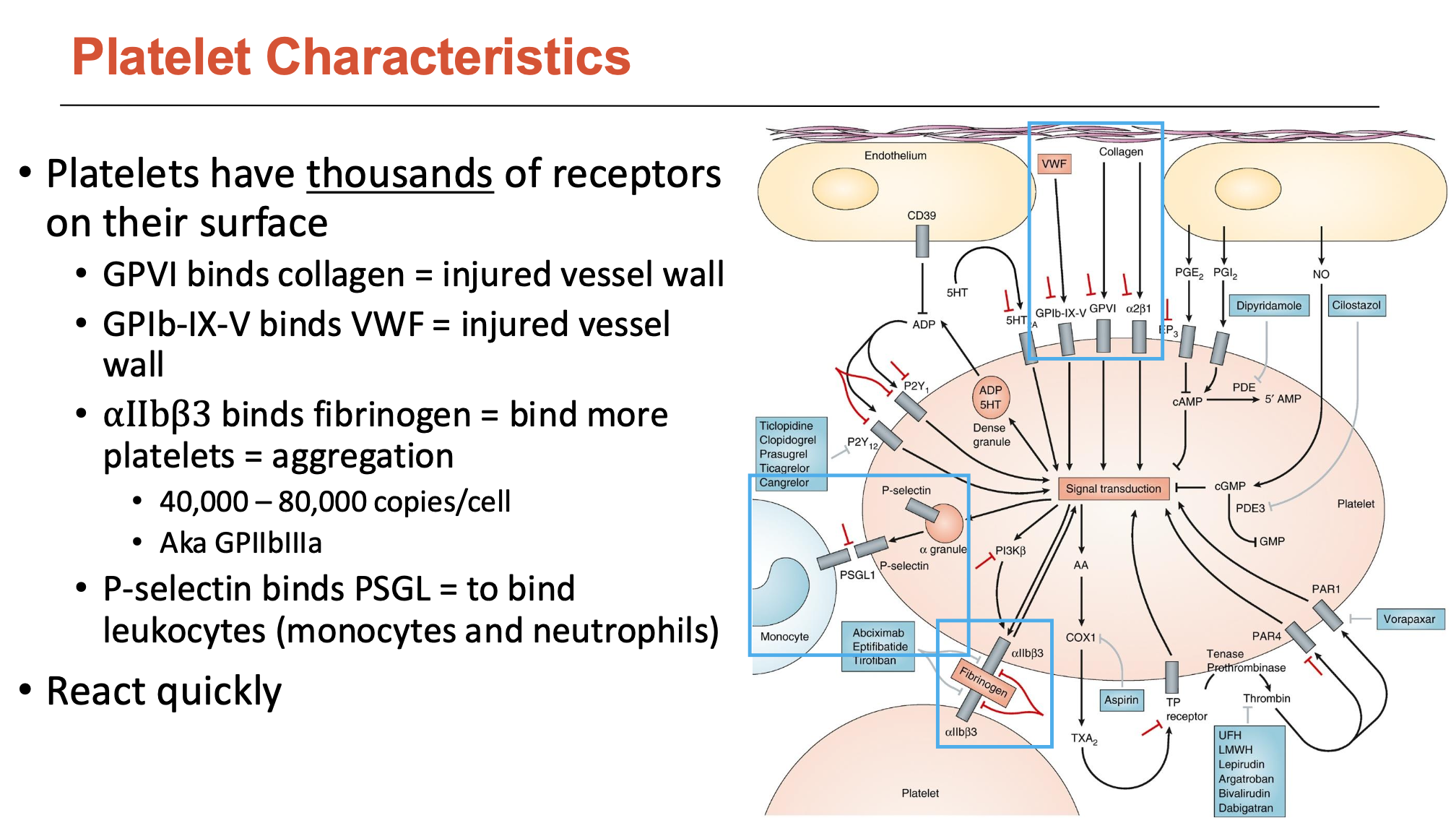
What do platelet receptors bind to (4)?
GP6 binds collagen = injured vessel wall
GPIb-IX-V binds VWF = injured vessel
wall
αIIbβ3 binds fibrinogen = bind more platelets = aggregation
40,000 – 80,000 copies/cell
Aka GPIIbIIIa
P-selectin binds PSGL = to bind leukocytes (monocytes and neutrophils)
What 3 factors are produced by the endothelium that contributes to thrombosis/coagulation? List examples
The endothelium produces inhibitors and activators of thrombosis/coagulation:
Anticoagulants
Thrombomodulin/Protein C
Sequesters ATIII on HS
Anti-platelets
Nitric oxide (NO)
PGI2
Factors to breakdown fibrin
tPA activated plasmin
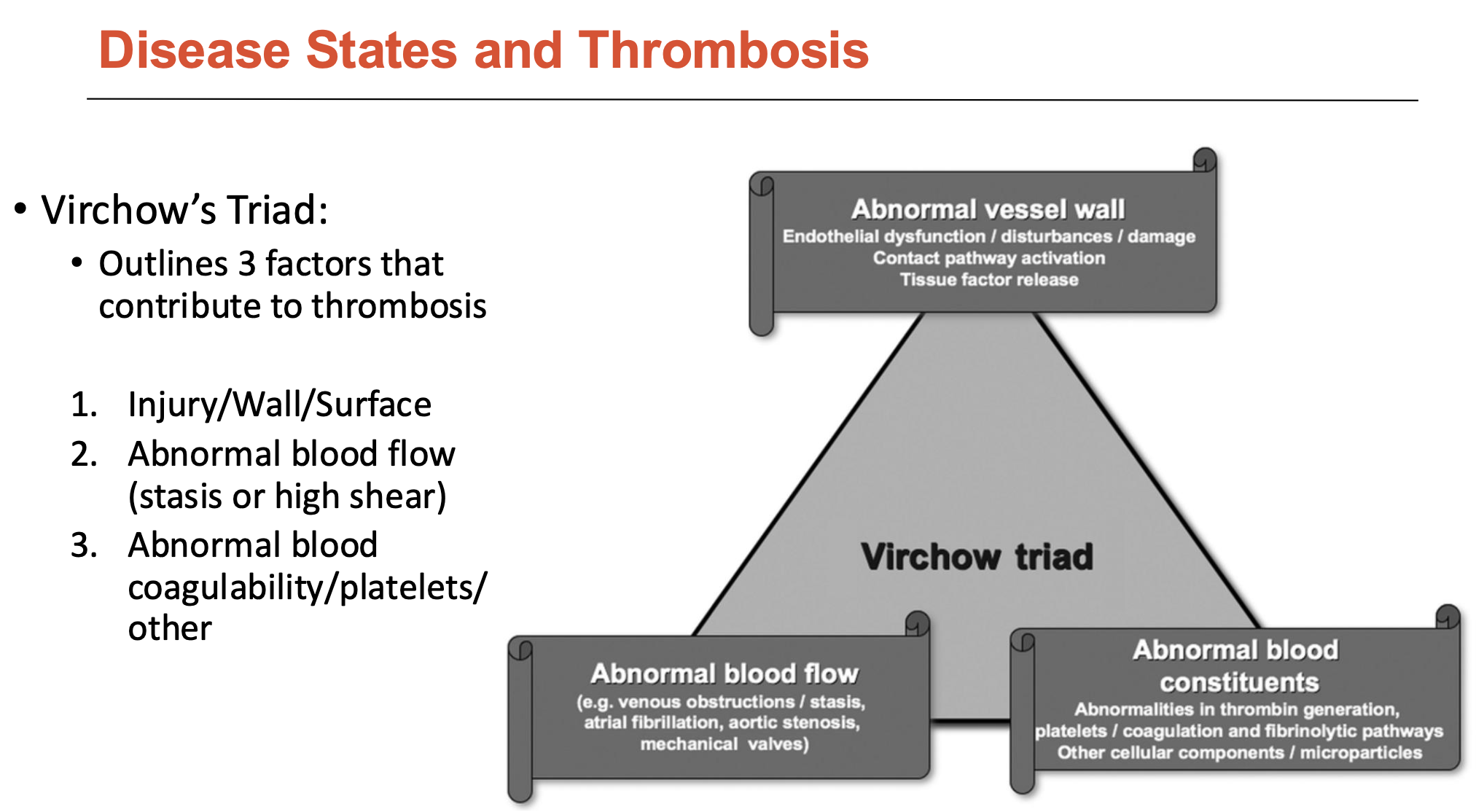
What is Virchow’s Triad?
Virchow’s Triad outlines 3 factors that contribute to thrombosis:
Injury/wall/surface
Abnormal blood flow (stasis or high shear)
Abnormal blood coagulability/platelets/other
List some examples of thrombosis in CVD
Ischemia-related thrombosis
arterial system
high flow (therefore high shear)
platelet-rich thrombi (white clots)
found in myocardial infarction, stroke, peripheral arterial disease
Venous thrombosis
venous system
low flow
fibrin-rich thrombi (red clots)
found in deep vein thrombosis
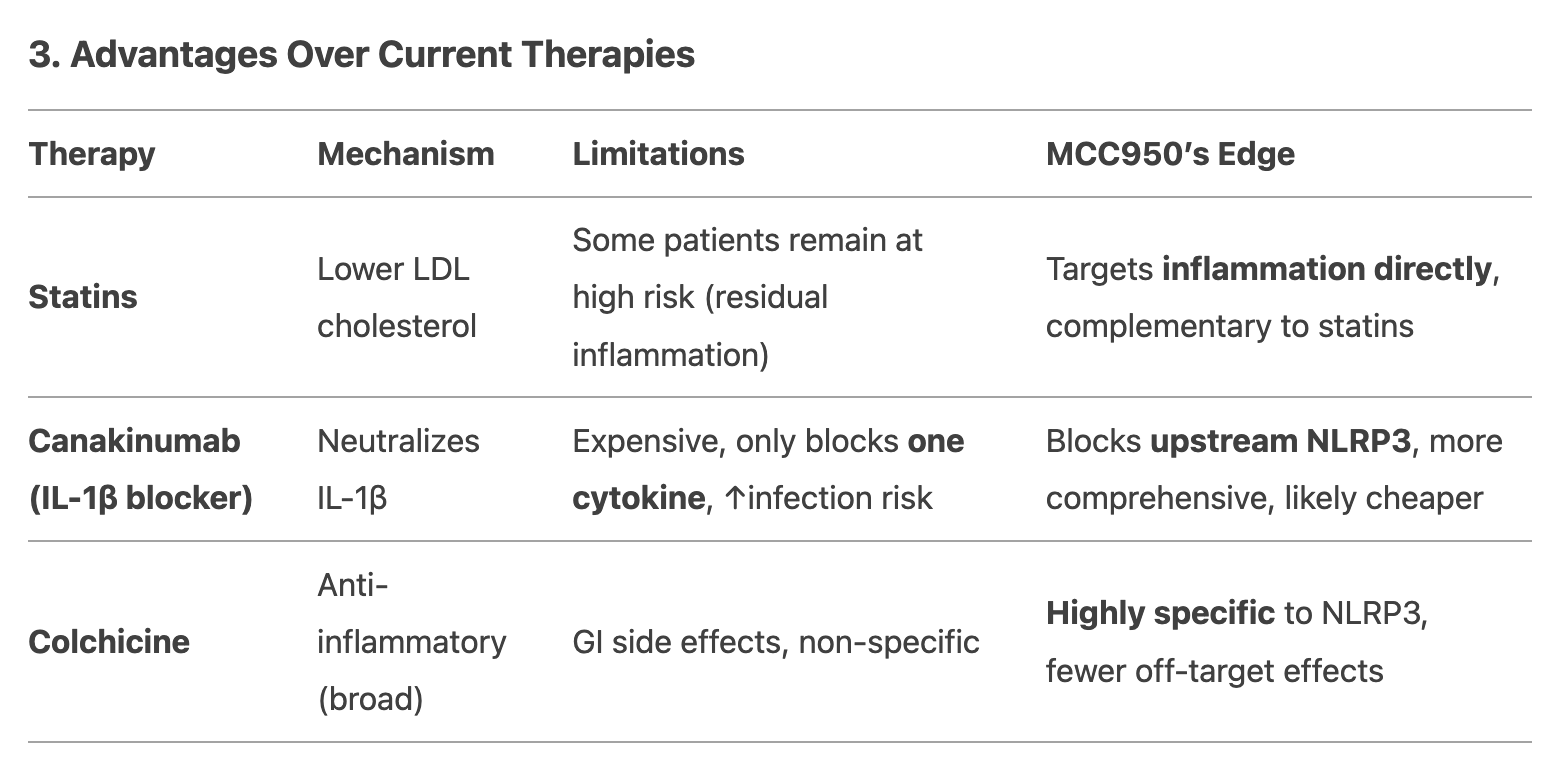
What happens in arterial-disease atherothrombosis?
Note: atherothrombosis is plaque rupture, atherosclerosis is plaque buildup
Narrowing of artery = stenosis
→ high shear (physical force)
→ shear gradient activates platelets and vWF
Thrombosis under laminar flow:
similar to platelet plug formation
Thrombosis under disturbed flow:
shear activation of vWF
shear activation of platelets leads to platelet adhesion and aggregation
accumulation of coagulation factors and degranulated platelets in the vortex/flow recirculation (deceleration) region
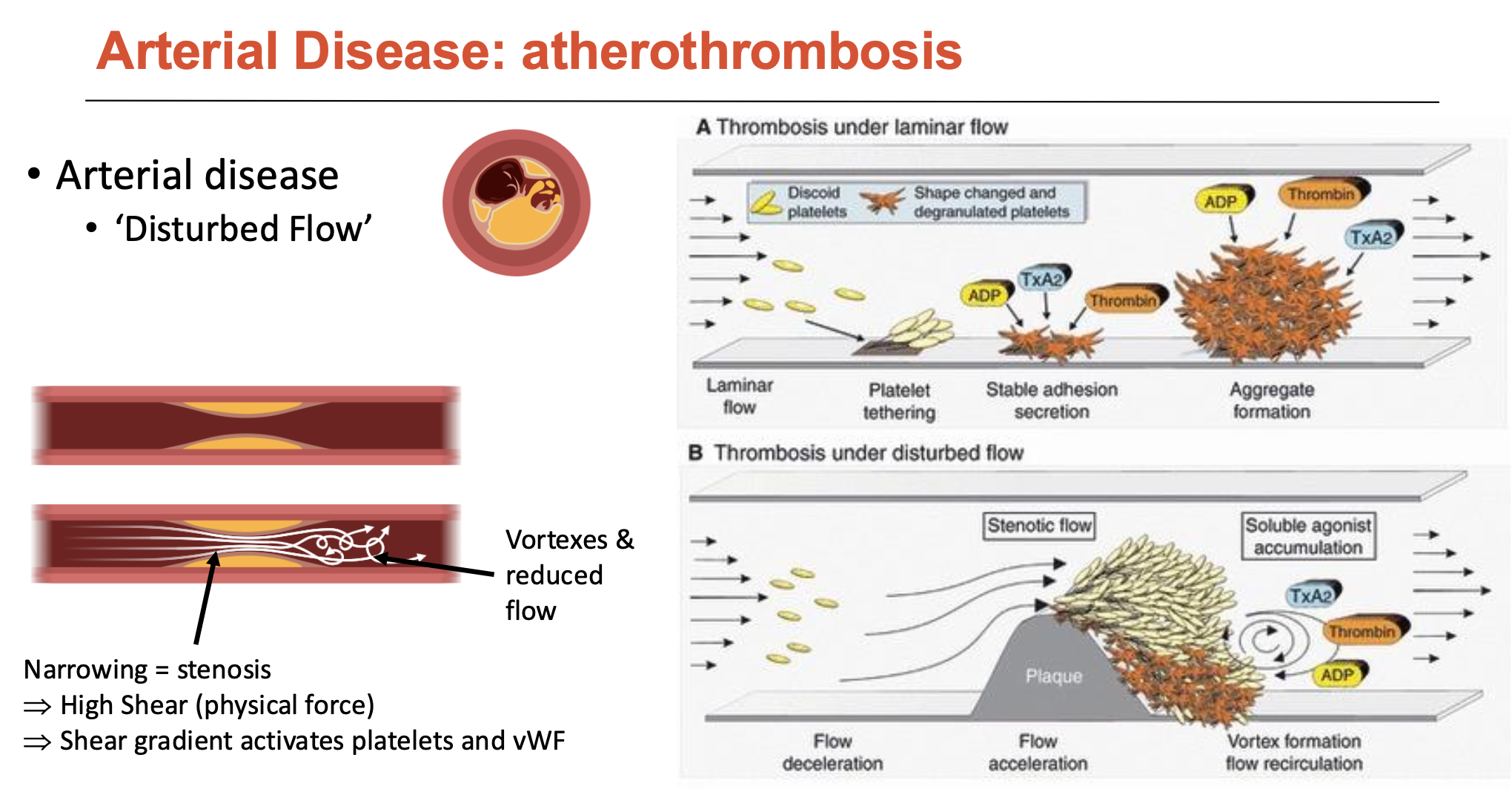
How is vWF shear-sensitive?
vWF is a globular protein (normally floats around blood as a compact blob, but can elongate)
can anchor to endothelium (via integrin αvβ3 or P-selectin)
vWF unfolds with high shear forces (high blood flow) → vWF strings
vWF strings can self-associate (end-to-end: 100μm to 1mm long)
vWF can be cleaved by ADAMTS 13
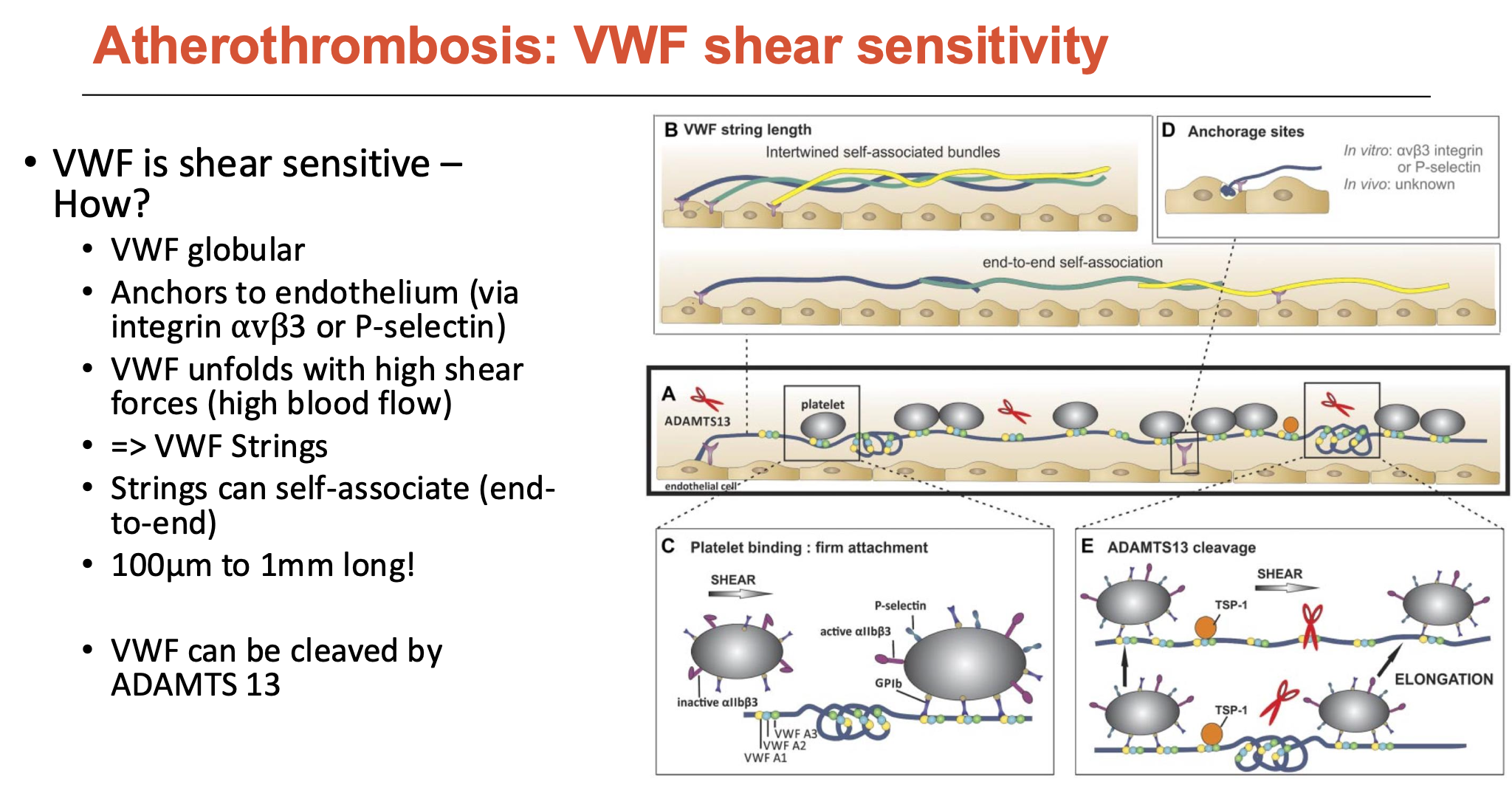
How are platelets shear-sensitive?
Platelets have many receptors, but intracellularly, many signals go through PI 3-Kinase (PI3K)
PI3K is an enzyme that phosphorylates things and triggers intracellular signalling to activate platelet roles
Specific types are shear-sensitive, and when blocked:
blocked PI3Kβ abolishes platelet-VWF binding at high shear (not at low shear)
blocked PI3KC2α increases reperfusion (without altering bleeding time)
How is revascularisation done for thrombosis (2)?
Revascularisation is the process of restoring blood flow by opening up blood vessels. Can be done by:
Breaking down the clot intravenously
tissue plasminogen activator (tPA) activates plasmin to cleave fibrin
but this has a very short window of therapeutic benefit
therefore, new drugs are in trials to improve this
Endovascular thrombectomy
remove the clot mechanically
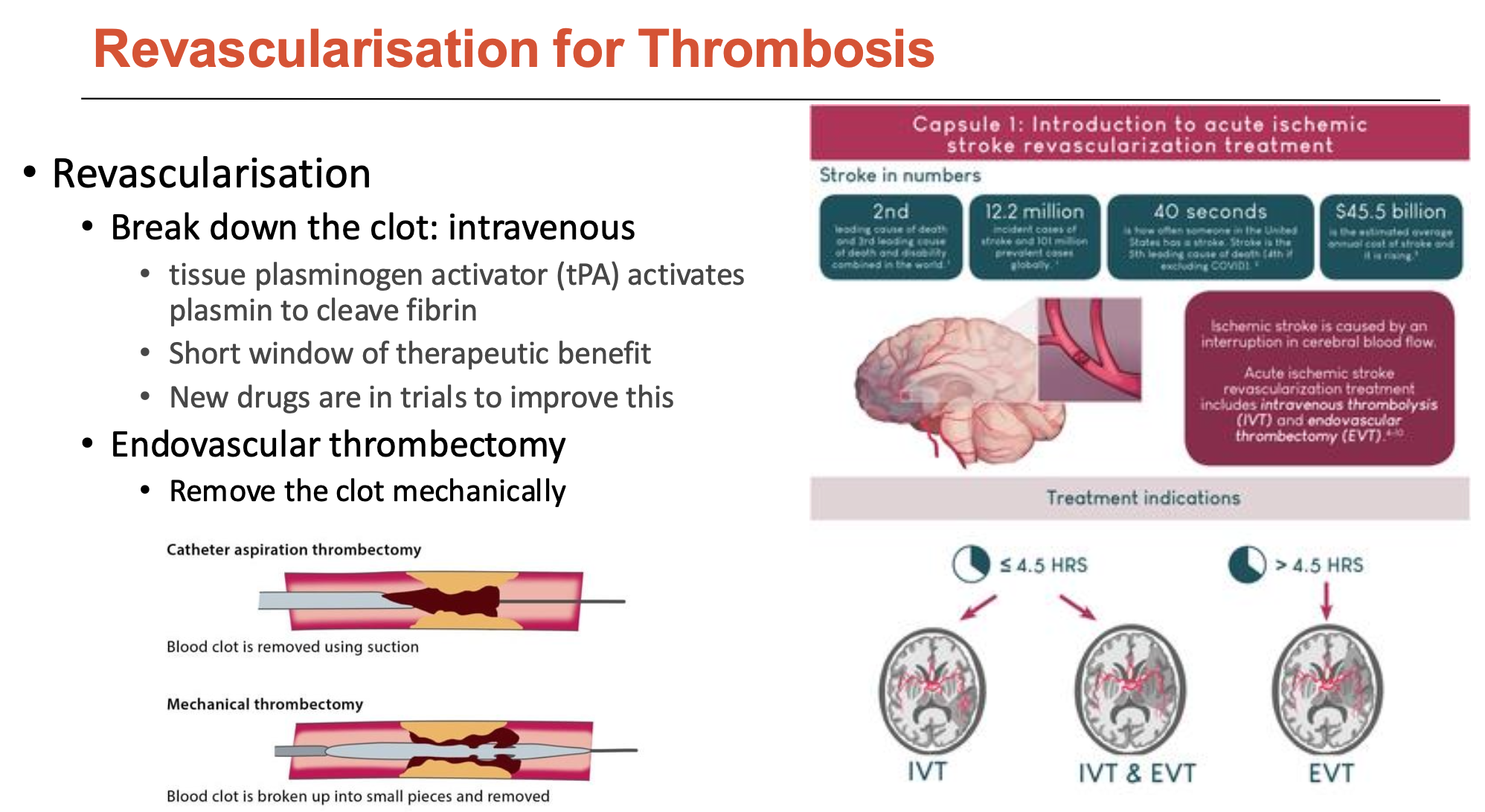
What are the most common anti-platelet drugs and what do they do?
Most common are:
P2Y1 or P2Y12 receptor inhibitors
Aspirin
These anti-platelet drugs:
block platelet adhesion/activation
aim to block thrombosis and not affect hemostasis
What are the long-term consequences of atherothrombosis?
Depends on severity and location, but more proximal to the heart = more tissue damage (due to downstream blood restriction).
Some examples of long-term consequences include:
electrical: arrhythmia
mitral valve prolapse (damaged papillary muscles)
heart failure
What happens in venous-disease atherothrombosis?
flow ‘stasis’ → induces endothelial dysfunction
antithrombin usually accumulates at the bottom of the valve, but endothelial dysfunction reduces antithrombin → instead, there is hypoxia in valve pocket
leads to accumulation of coagulation factors
How can venous thrombosis be managed?
Acute venous thrombosis:
dissolve thrombus
thrombolytic (tissue plasminogen activator = tPA)
Managing long-term venous thrombosis:
anticoagulants
UFH = unfractionated heparin
LMWH = low molecular weight heparin
Warfarin = vitamin K antagonist (blocks coagulation factor production in liver)
DOAC = direct oral anticoagulant (Fxa or thrombin inhibitor)
Overall, aim is to balance anticoagulation with bleeding
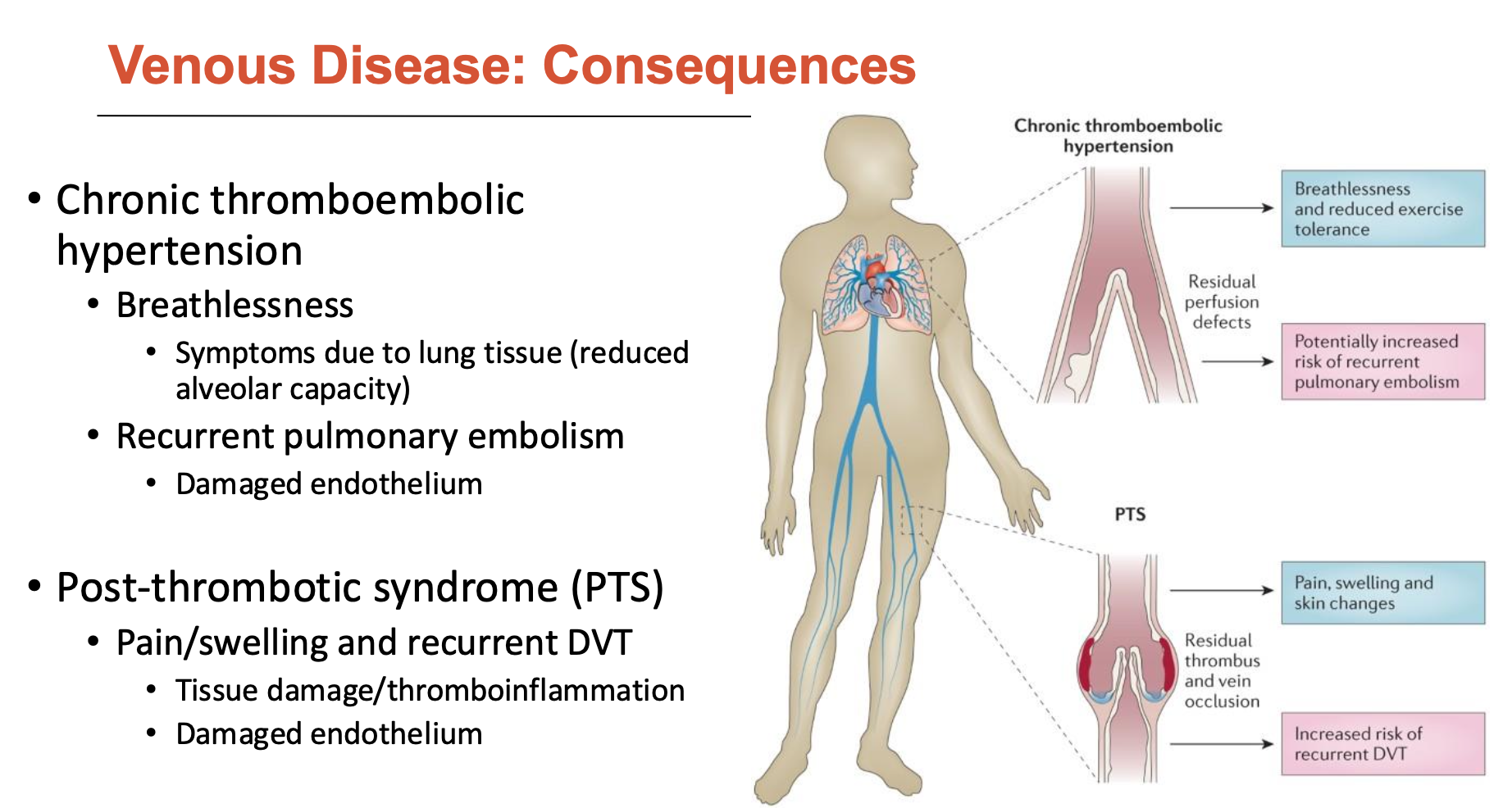
What are the consequences of venous disease?
Chronic thromboembolic hypertension
Breathlessness: due to lung tissue (reduced alveolar capacity)
Recurrent pulmonary embolism: damaged endothelium
Post-thrombotic syndrome (PTS)
pain/swelling and recurrent DVT
tissue damage/thromboinflammation
damaged endothelium
What roles do platelets have in the immune system?
Platelets produce cytokines and microparticles (inflammation)
But, main role is in innate and adaptive immunity: host defence
Innate:
directly sense pathogen via pseudopodia/toll-like receptor (TLR)
neutrophil, monocyte
cause NET formation
phagocytose microorganisms (phagosome-like vacuoles)
Adaptive:
antigen-presenting cell (APC)
T and B cells
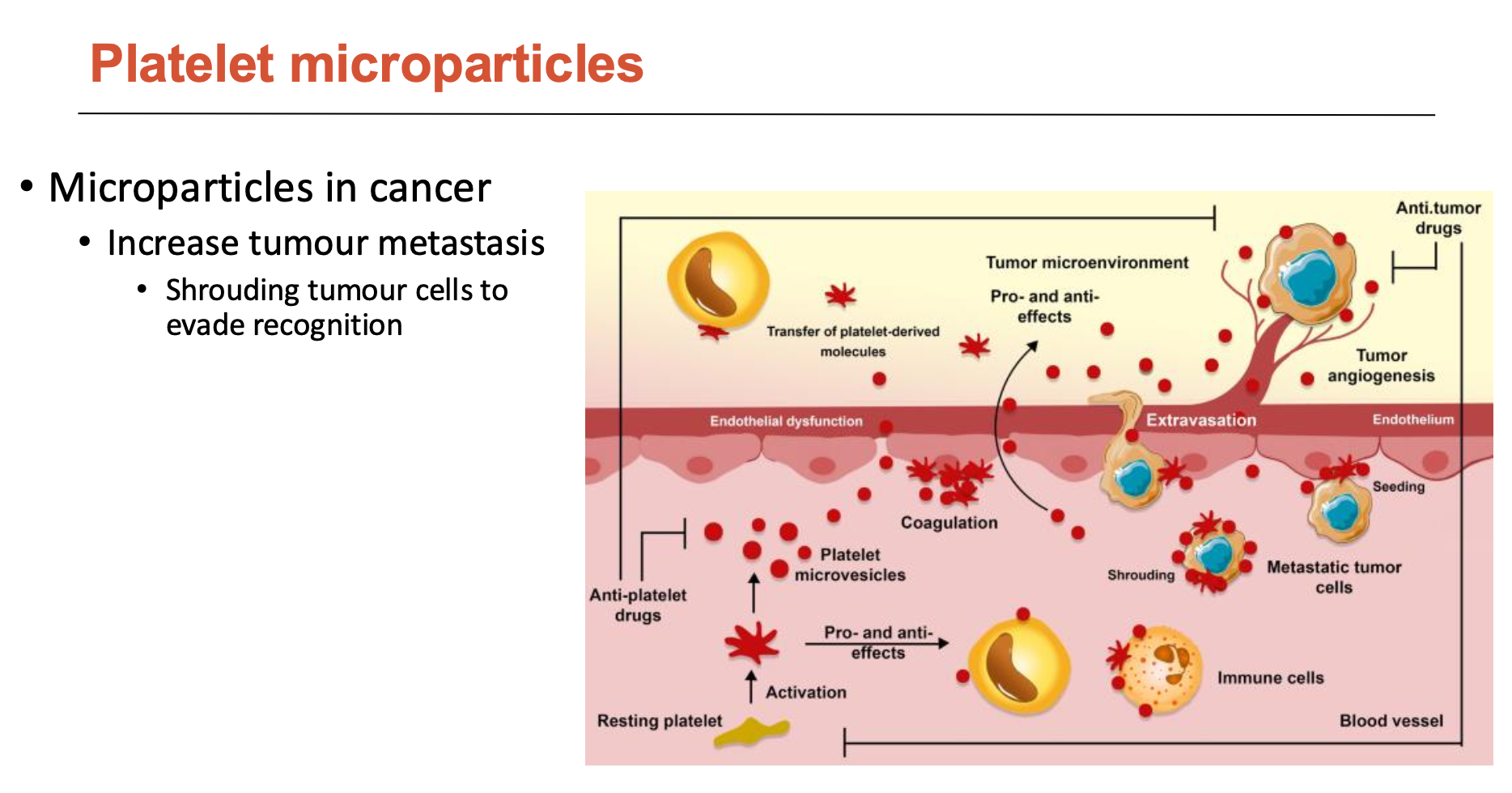
What is the features of platelet microparticles?
Microparticles play a role in:
coagulation, thrombosis, and thromboinflammation
pro- and anti-inflammatory effects - scan vasculature
support angiogenesis
blocking microlesions at sites of leukocyte extravasation/transmigration
increase tumour metastasis
shroud tumour cells to evade recognition
Evaluate Coronary Artery Bypass for CAD
Coronary artery bypass is repair made using vessels from patient: either vein from leg (SV) or artery from chest (IMA)
IMA is significantly more effective but not always available
No commercial vascular graft can be used in this setting
CAD is highly invasive → long recovery times and significant morbidity
especially since most CAD patients are old and sick → higher risk
Evaluate Percutaneous Coronary Intervention (PCI or Angioplasty) for CAD
Alternative to coronary artery bypass, PCI is a non-invasive day surgery
generally femoral artery access, but can be radial
live x-ray (angiography) used to identify blockages and place balloon
however, blockages in mild disease can be subtle and the link between angiogram and lesion is not always clear
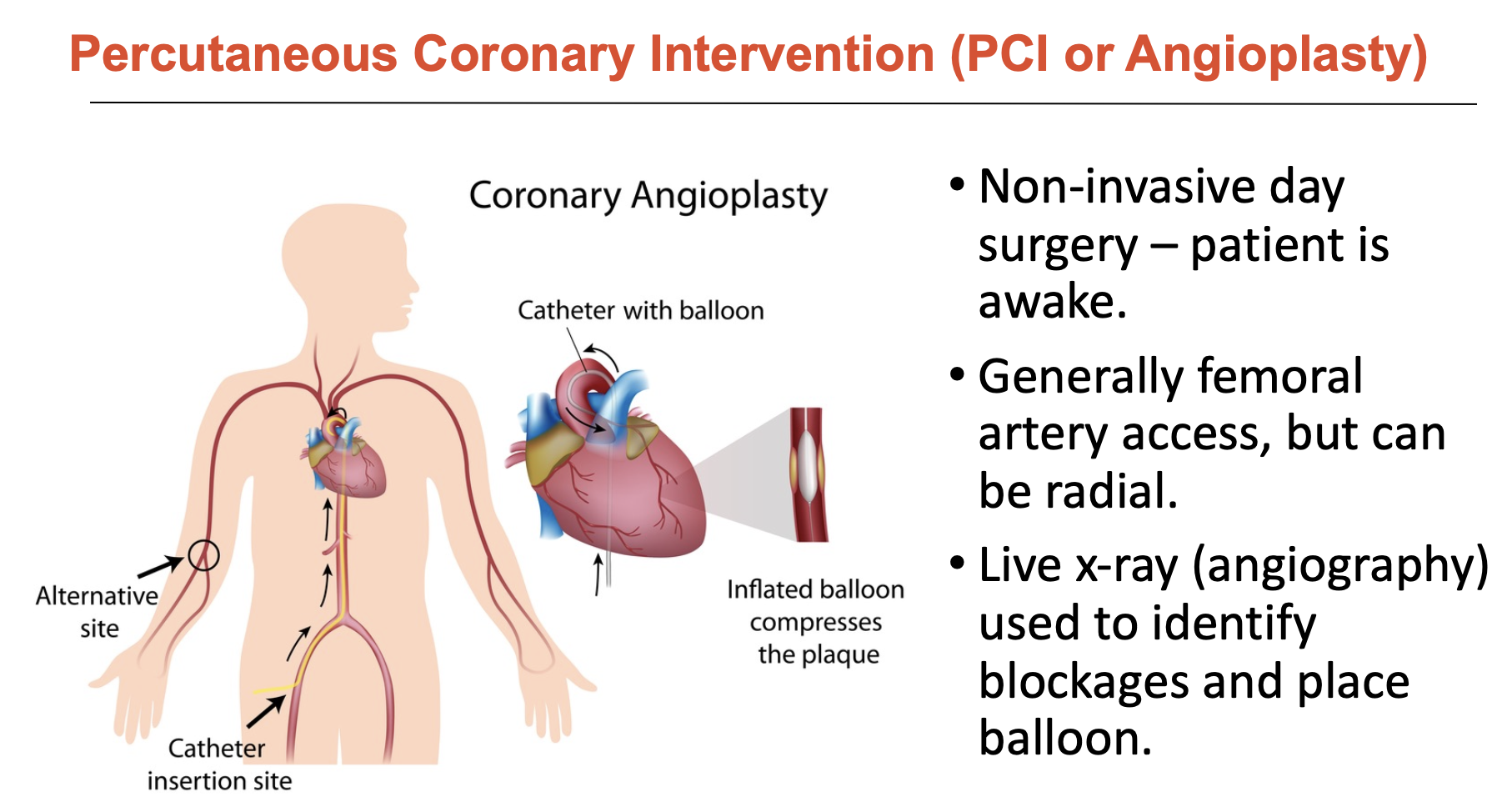
What was balloon angioplasty?
Balloon angioplasty was used to identify blocked areas since angiography is hard to rely upon accurately.
deflated balloon is advanced to lesion site → expanded at high pressure to push back and compress plaque
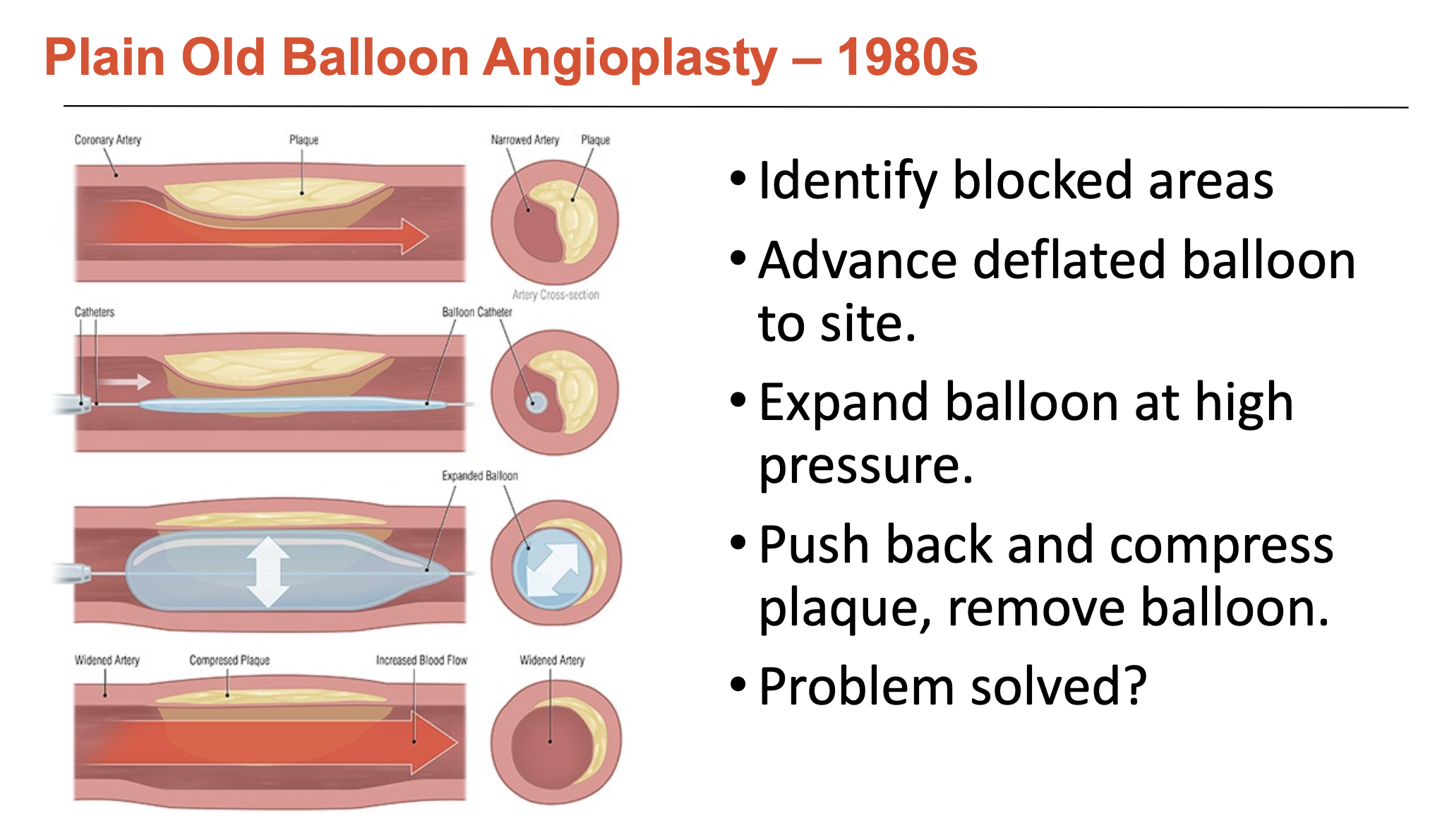
What issues were associated with balloon-only angioplasty (3)?
Abrupt closure:
balloon injury can severely damage vessel wall, the high pressure creates ’flaps’ that block the blood flow
occurs in 5-10% patients; 20% of these require emergency bypass surgery
Endothelial damage:
Angioplasty severely damages the endothelium
high-pressure balloon expansion removes most of this protective layer
leaves behind a highly inflammatory, thrombogenic environment
Severe re-narrowing
Activation of immune cells, which subsequently activate smooth muscle cells
SMCs migrate from media, proliferate, secrete proteins, glycoproteins, and proteoglycans
30-50% of patients at 6 months
What is coronary stenting?
Like balloon angioplasty but using a small, expandable mesh tube to keep artery open
stents can only be made of a few handful of metals
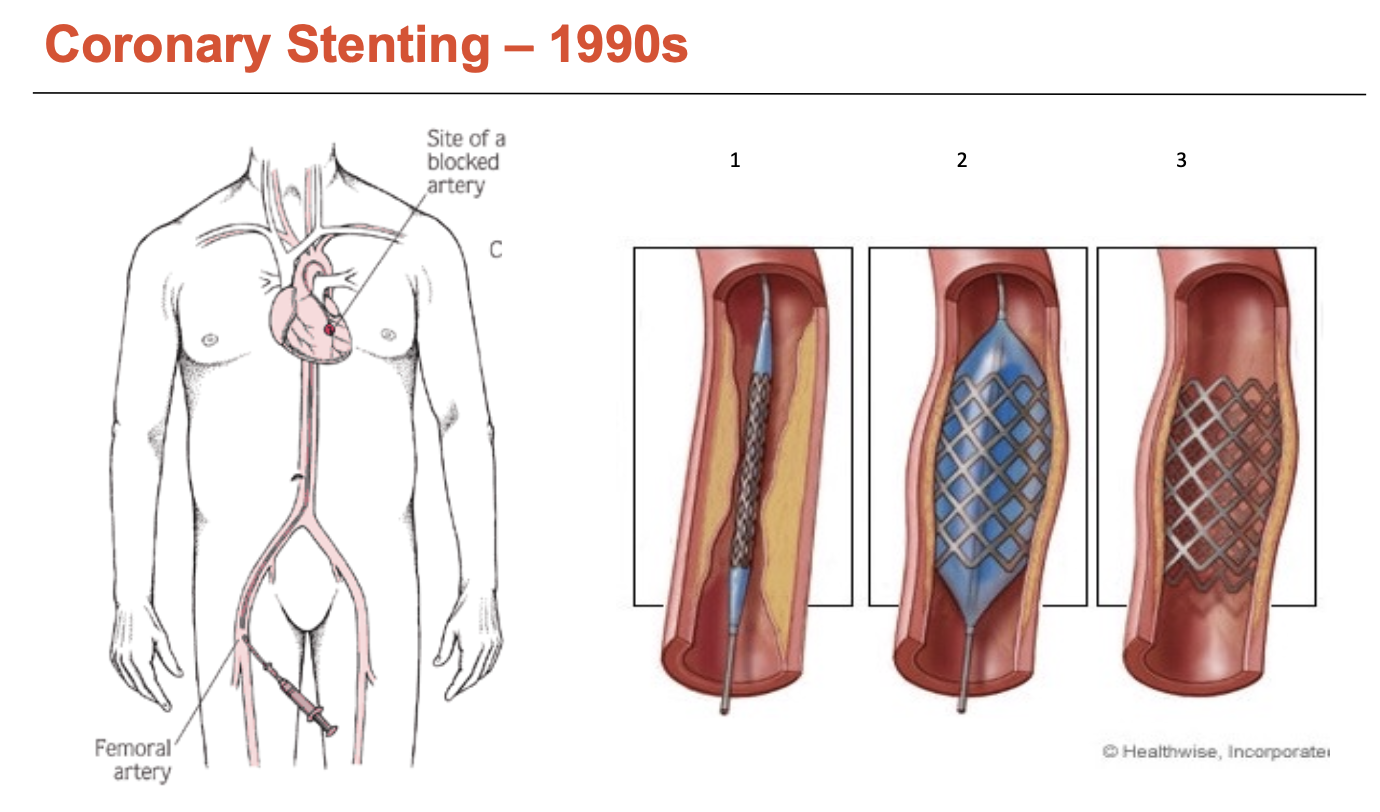
What are the issues with bare-metal coronary stents (3)?
Stents damage endothelium
While it does reduce vessel recoil, it still removes most of the local endothelial layer (similar to balloon angioplasty)
It penetrates the arterial wall, damaging protein matrix, → induces inflammation
human endothelial recovery takes months (6-7 months for full repair)
Thrombosis
Blockage is sudden and total (increased risk of mortality)
Up to 25% experienced early stent thrombosis (<30 days) → a further 25% among these die
Patients require medication (anti-platelet + aspirin) for 6-12 months - called DAPT (dual anti-platelet therapy - drops thrombosis rates to 1-2%)
DAPT duration is a significant concern
In-stent restenosis
Affects 30-50% of patients
Driven by vessel recoil, EC damage, ongoing inflammatory response
occurs in 3-9 months, required re-intervention (driven by SMC proliferation)
What are the key drug considerations for coronary stenting (2)?
Sirolimus interferes with cell cycle at G1 → stops cells dividing
Paclitaxel is poisonous
What are drug-eluting stents?
Stents coated with drugs that inhibit cell division (sirolimus and paclitaxel), which aimed to target the re-stenosis issue
What were the issues with drug-eluting stents (3)?
Late Catch-Up
Drug has to wear out at some point
Inhibition of SMCs is not permanent and regrowth was seen within a few years (late stent thrombosis)
drug-elution kills endothelium as well as SMCs → delays healing
Overlapping Stents
Increase inflammation and fibrin (clot)
Slower re-endothelialisation
Off-target effects
on progenitor cells and activation of the clotting cascade → significant increase in mortality
How did second generation drug-eluting stents evolve (2)?
stronger metal allows thinner struts
analogues of sirolimus aimed to do less damage
More effective → demonstrated less restenosis and inflammation, lower rates of thrombosis, death, and MI
BUT restenosis and thrombosis still persist (even at lower rates)
How is heart function measured?
Epicardiography can be used to observe heart function
Heart function can be measured as the ejection fraction: EF (%)
= [(end-diastolic volume - end-systolic volume) / end-diastolic volume] x 100
= [(stroke volume) / (end-diastolic volume)] x 100
Healthy EF = between 50 to 70%
![<p>Epicardiography can be used to observe heart function</p><p>Heart function can be measured as the <strong>ejection fraction: </strong>EF (%) </p><p>= [(end-diastolic volume - end-systolic volume) / end-diastolic volume] x 100</p><p>= [(stroke volume) / (end-diastolic volume)] x 100</p><p></p><p>Healthy EF = between 50 to 70% </p><p></p>](https://knowt-user-attachments.s3.amazonaws.com/d21661f6-6db0-450f-8bba-374ef4b14e68.png)
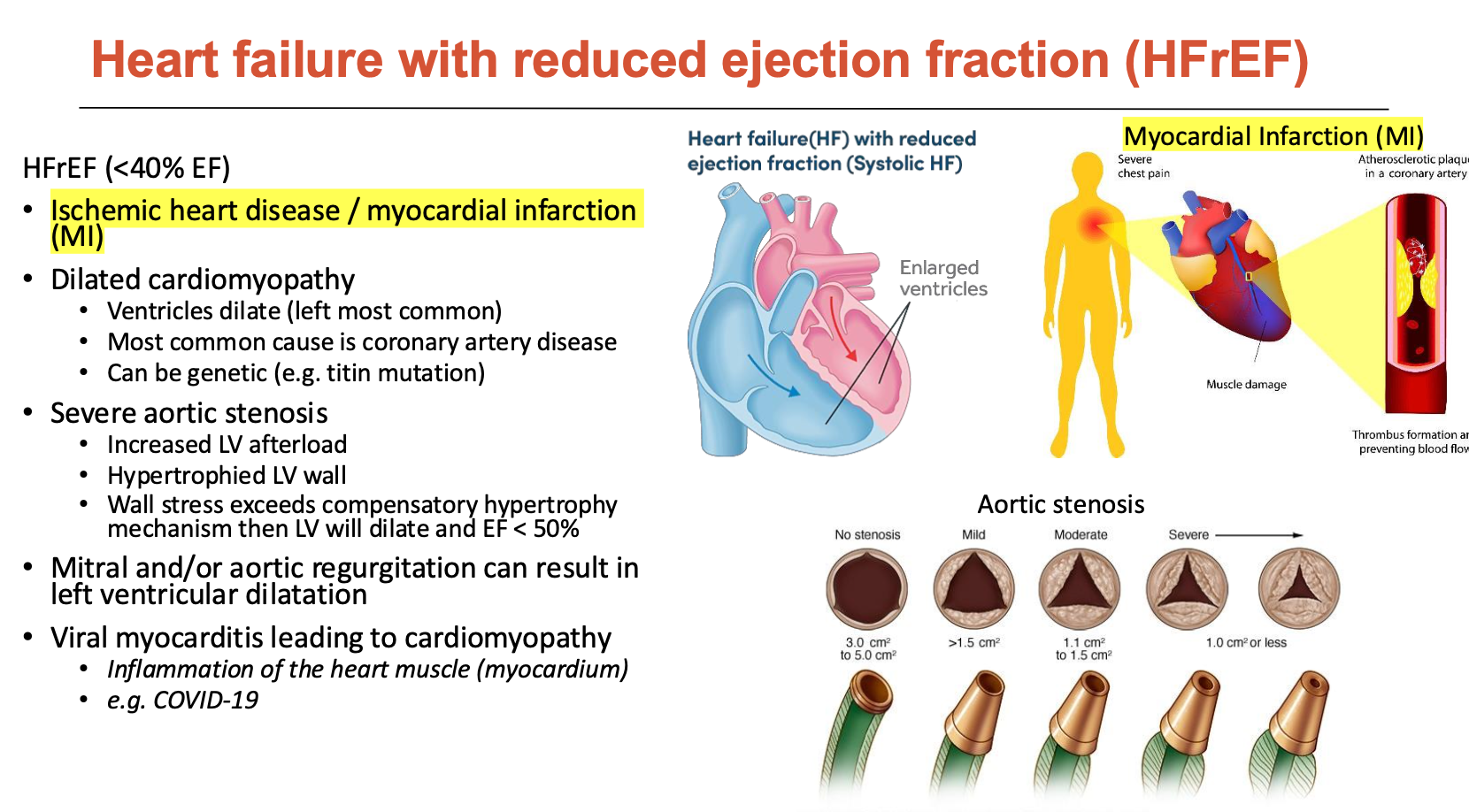
What is heart failure with reduced ejection fraction?
HFrEF is when EF < 40%
This is mainly due to ischemic heart disease/MI
How do different forms of heart failure lead to HFrEF (5)?
Ischemic heart disease / MI
heart muscle does not get enough oxygen → non-contractile tissue
increases EDV (LV dilation)
ESV increases even more so (reduced contraction) - we want ESV to be low
Dilated cardiomyopathy
ventricles dilate (left most common)
most common cause is CAD
can be genetic (e.g. titin mutation
Severe aortic stenosis
increased LV afterload
hypertrophies LV wall
wall stress exceeds compensatory hypertrophy mechanism then LV will dilate and EF < 50%
Mitral and/or aortic regurgitation can result in LV dilation
Viral myocarditis leading to cardiomyopathy
Inflammation of the heart muscle (myocardium) (e.g. COVID-19)
What happens to the heart post-MI?
Within minutes after MI, cardiomyocytes start dying and undergo coagulative necrosis
starts releasing DAMPs (danger-associated molecular patterns)
fibroblasts and macrophages also start releasing cytokines into the bloodstream → recruits other immune cells
Within a day, there is a massive influx of pro-inflammatory monocytes and neutrophils → some of the monocytes can differentiate into M1 (pro-inflammatory) macrophages
both will release a lot of MMPs → degrades ECM
macrophages will also phagocytose dead CMs
Within a few days, a lot of monocytes are being recruited and fibroblasts (responsible for collagen) increase in numbers
fibroblasts undergo a lot of proliferation
some even differentiate into myofibroblasts → produce even more collagen
monocytes are differentiating into M2 macrophages → more repair (building scar)
Within weeks, a lot of scar tissue is formed, while CMs do not proliferate
CMs are replaced with scar tissue
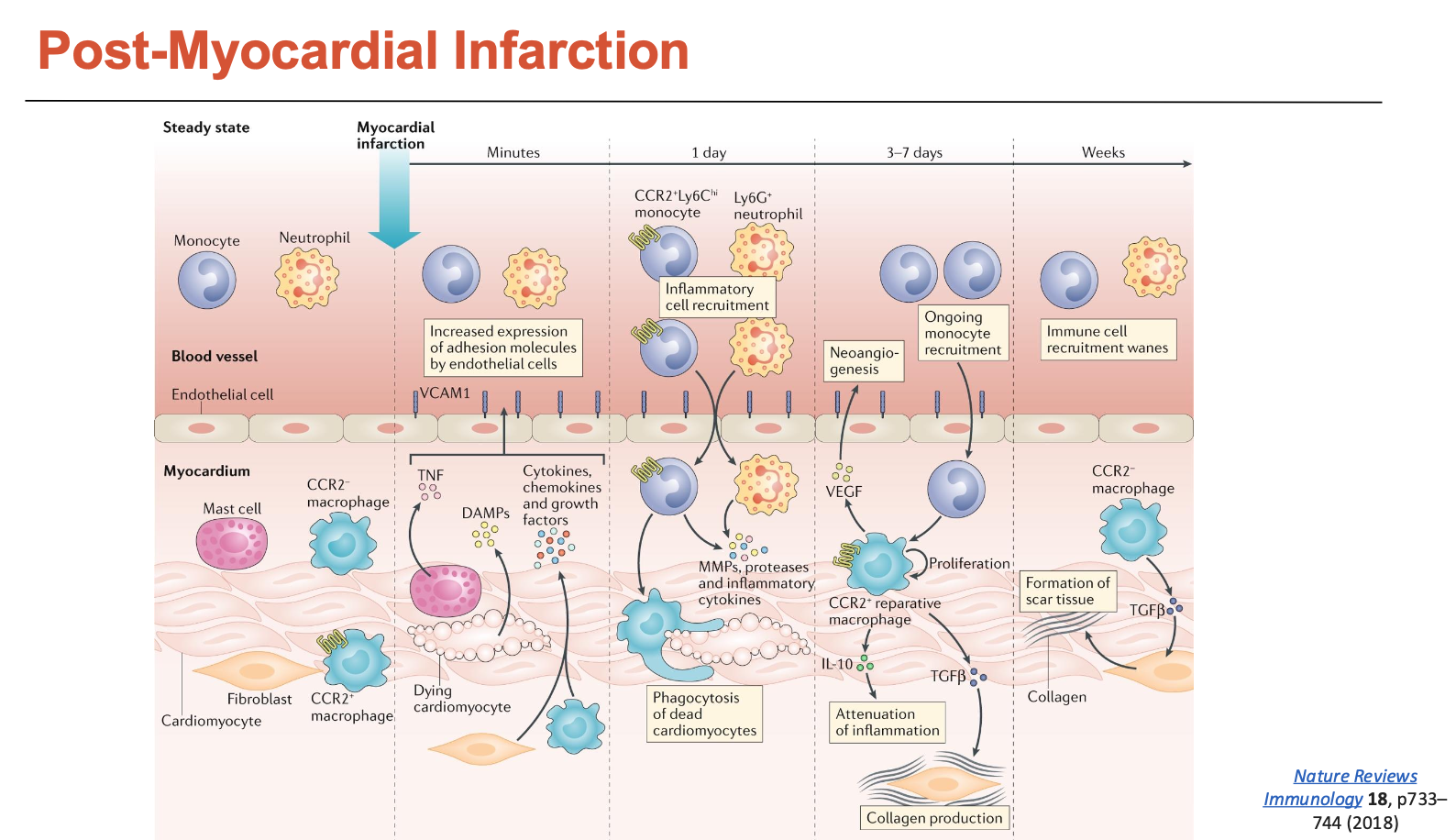
What are some post-myocardial infarction therapies?
Early intervention
place a stent to open up coronary arteries
most important (within 4 hours ideally)
stent can lead to ROS formation → reperfusion injury
Anti-inflammatories
Decreasing IL1 and IL6 can increase M2 macrophages and remodelling in animal models (good)
BUT, the same benefits are not shown in humans
Anti-fibrotics
Some fibrosis is required to avoid thin walls and risk of rupture
But, too much fibrosis negatively affects heart function
anti-fibrotics block myocardial differentiation to reduce scar tissue size (good in animal models, but these benefits not found in humans)
Induced pluripotent stem cells (iPSCs)
Derived from other tissue in the patient, iPSCs can be differentiated into various cell types → form new tissue to replace damaged tissue
Have been used in clinical trials successfully
What is heart failure with preserved ejection fraction (HFpEF)?
HFpEF is a problem with heart relaxation (diastolic dysfunction)
thickened LV wall (fibrosis) → stiffer → cannot relax
systemic inflammation
makes up >50% of heart failure (more prevalent than HFrEF)
heterogeneous disease, but age is a strong risk factor
female, obesity, hypertension, endothelial dysfunction, lung congestion
overlaps with other diseases: obesity, AF, kidney disease, sleep apnoea, diabetes