Understanding Hemoglobinopathies and Their Evaluation
1/156
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
157 Terms
What are hemoglobinopathies?
Hemoglobinopathies are defined as quantitative or qualitative abnormalities of globin synthesis caused by inherited mutations in the globin genes.
What is the difference between hemoglobinopathies and thalassemias?
All thalassemias are hemoglobinopathies, but not all hemoglobinopathies are thalassemias. Thalassemia specifically refers to diminished production of globin.
What type of inheritance is associated with thalassemias?
Thalassemias are inherited in an autosomal recessive manner.
What are the two types of hemoglobin abnormalities?
The two types of hemoglobin abnormalities are quantitative defects (imbalance of chain synthesis) and qualitative defects (additions, substitutions, or deletions of amino acids).
What are examples of quantitative defects in hemoglobin?
Examples of quantitative defects include thalassemia syndromes, specifically alpha and beta thalassemia.
What are examples of qualitative defects in hemoglobin?
Examples of qualitative defects include HbD, HbC, and sickle cell disease.
What mutation causes sickle cell disease?
Sickle cell disease is caused by a mutation where GAG is replaced by GTG, resulting in the substitution of Glu with Val.
What are the alpha globin genes associated with?
Alpha globin genes are located on chromosome 16.
What are the beta globin genes associated with?
Beta globin genes are located on chromosome 11.
What is the normal adult hemoglobin structure?
Normal adult hemoglobin is composed of two alpha and two beta chains, represented as a2b2.
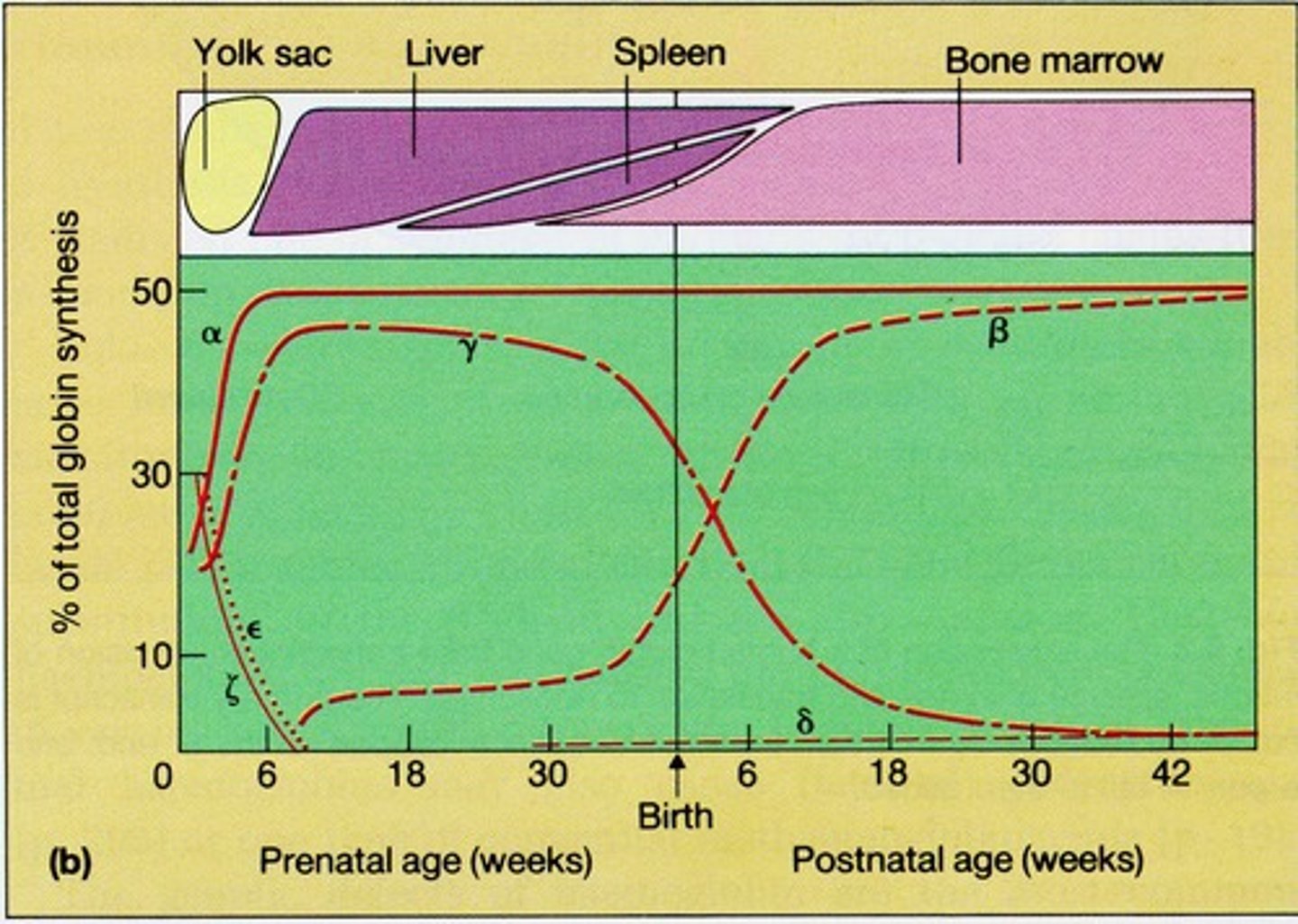
What is fetal hemoglobin composed of?
Fetal hemoglobin is composed of two alpha and two gamma chains, represented as a2g2.
What happens during hemoglobin switching?
Hemoglobin switching refers to the transition from fetal hemoglobin (HbF) to adult hemoglobin (HbA1) as a person ages.
What is the significance of equal numbers of alpha and beta chains?
Equal numbers of alpha and beta chains are needed for normal hemoglobin function.
What occurs in thalassemias due to chain imbalance?
In thalassemias, excess beta chains can form a beta tetramer (HbH), and excess gamma chains can form a gamma tetramer (HbBarts), both of which are poor oxygen transporters.
What is the role of a complete blood count (CBC) in evaluating hemoglobinopathies?
A CBC evaluates the number of red blood cells, hemoglobin levels, and mean corpuscular volume (MCV) to assess for abnormalities.
What does the mean corpuscular volume (MCV) indicate?
The mean corpuscular volume (MCV) indicates the average size of red blood cells and can suggest conditions like iron deficiency.
What does mean corpuscular hemoglobin (MCH) measure?
Mean corpuscular hemoglobin (MCH) measures the average amount of hemoglobin in red blood cells.
What is the life span of red blood cells?
Red blood cells have a life span of approximately 120 days.
How many trips do red blood cells make through the heart during their life span?
Red blood cells make about 500,000 trips through the heart.
How many trips do red blood cells make through the spleen?
Red blood cells make about 10,000 trips through the spleen.
What is the total distance traveled by red blood cells during their life span?
Red blood cells travel a total distance of approximately 186 miles (316 km).
What is the primary function of hemoglobin in red blood cells?
The primary function of hemoglobin is to carry oxygen to body tissues.
What does RDW stand for and what does it indicate?
Red blood cell distribution width; it indicates the range of variation of RBC volume.
What is HPLC and its role in evaluating hemoglobinopathies?
High performance liquid chromatography; it separates, identifies, and quantifies each component in a blood sample.
What are the three types of hemoglobin measured in HPLC for thalassemia evaluation?
HbA (adult hemoglobin α2β2), HbF (fetal hemoglobin α2γ2), and HbA2 (delta globin α2δ2).
What is the normal range for HbA in adults?
Greater than 96%.
What does it indicate if the total hemoglobin components do not add up to 100%?
There may be an unidentified variant present that requires further investigation.
How do HPLC and hemoglobin electrophoresis differ?
They are often used interchangeably, but they are not the same.
What is the reference range for RBC count in a normal adult?
4.20-5.80 million cells/mcL.
What is the significance of MCV in hemoglobinopathy evaluation?
Mean corpuscular volume (MCV) helps in determining the size of red blood cells and can indicate types of anemia.
What does a low MCV and MCH indicate in a blood test?
It suggests microcytic hypochromic anemia, often associated with iron deficiency or thalassemia.
What are the normal ranges for HbA2 and HbF in adults?
HbA2: 1.8-3.2%, HbF: less than 2%.
What does an elevated HbA2 level (>3.5%) suggest?
It may indicate beta thalassemia.
What is the normal range for hemoglobin in adults?
13.2-17.1 g/dL.
What are the hemoglobin fractions for a newborn child?
HbA1: 20-30%, HbA2: 0.2%, HbF: 70-80%.
What is the significance of checking HbA2 levels in thalassemia carriers?
Elevated HbA2 levels can indicate beta thalassemia, while normal levels may suggest alpha thalassemia.
What is the role of ferritin in evaluating iron status?
Ferritin measures stored iron and helps assess if the body is storing adequate iron.
What populations are commonly affected by alpha thalassemia?
Southeast Asians, Chinese, Mediterranean, Middle Eastern, and African American populations.
What is the reference range for RDW in a normal adult?
11.0-15.0%.
What does a high RDW indicate?
It can indicate variability in red blood cell size, often seen in various types of anemia.
What are the implications of a patient having both low MCV and elevated HbA2?
It may suggest the presence of beta thalassemia trait.
What is the significance of identifying thalassemia carriers?
It helps in genetic counseling and managing potential health risks.
What is the importance of performing a CBC in hemoglobinopathy evaluation?
A complete blood count (CBC) helps identify abnormalities in red blood cell size, hemoglobin concentration, and overall blood health.
What is the reference range for MCH in adults?
27.0-33.0 pg.
What are the common populations where Alpha Thalassemia is identified?
Northern Europeans, Egyptians, South Africans, Asian Indians, Saudi Arabians, and Japanese (rare).
What tests are used to identify Alpha Thalassemia carriers?
Hemoglobin electrophoresis/HPLC, Quantitative Hb A2 (normal), Quantitative Hb F (normal to slight elevation), CBC (low MCV, MCH, high RDW), and FEP (or other iron studies).
What is the significance of cis vs trans Alpha Thalassemia trait?
Cis Alpha Thalassemia trait has more clinical significance than trans Alpha Thalassemia trait.
What is the typical mRNA ratio of a2 to a1 in individuals with a normal alpha globin genotype?
In individuals with a normal (aa/aa) alpha globin genotype, a2 mRNA is present at an average 2.8-fold excess to a1.
What is the most common deletion in Alpha Thalassemia?
Alpha 3.7 Deletion.
What causes the Alpha 3.7 Deletion?
It is caused by unequal crossover between chromosomes at meiosis.
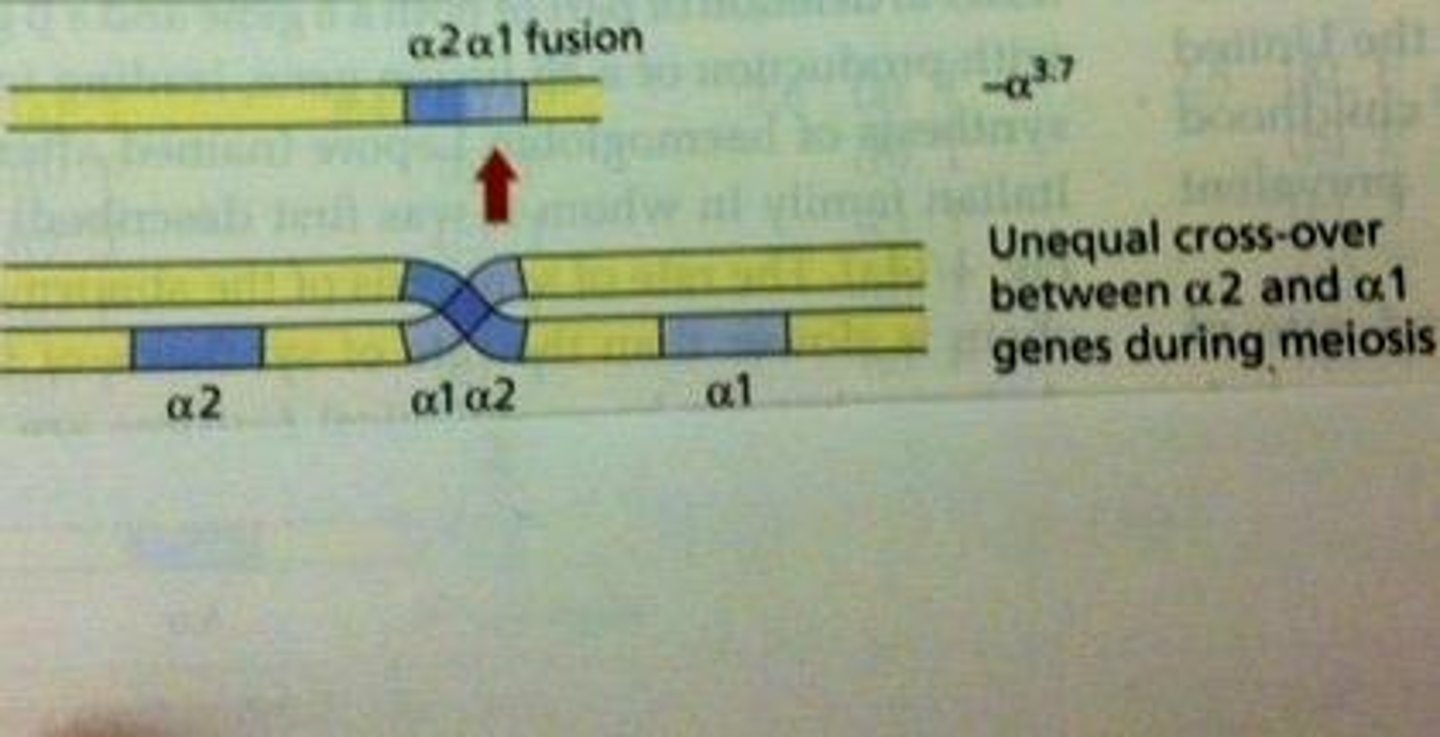
What is the clinical significance of the Alpha 3.7 Deletion?
It is often a 'silent carrier' and can lead to mild anemia.
What are the four types of Alpha Thalassemia based on the number of genes deleted?
1 gene deletion: silent carriers; 2 gene deletion: trait/minor; 3 gene deletion: Hb H disease; 4 gene deletion: hydrops fetalis (incompatible with life).
What is Hemoglobin H disease?
It occurs when 3 of the 4 genes are deleted, causing an imbalance between alpha and beta chain production, leading to excess beta chains and abnormal hemoglobin.
What is the effect of point mutations in Alpha Thalassemia?
Point mutations can lead to conditions like Hemoglobin Constant Spring, which results in an unstable elongated protein.
What is the mutation associated with Hemoglobin Constant Spring?
A T→C mutation (TAA→CAA) in codon 142 that results in an unstable protein 31 amino acids longer.
What is the prevalence of Hemoglobin Constant Spring?
It has a high incidence in Southeast Asians and Chinese.
What are the symptoms of Hemoglobin H disease?
Life-threatening symptoms due to the destruction of RBCs and poor oxygen carrying capacity.
What is the typical presentation of individuals with 2 gene deletions in Alpha Thalassemia?
They may present with mild anemia.
What is the expected outcome for individuals with 4 gene deletions in Alpha Thalassemia?
Hydrops fetalis, which is incompatible with life.
What is the role of DNA analysis in diagnosing Alpha Thalassemia?
Alpha Thalassemia is an exclusion diagnosis unless DNA analysis is utilized.
What is the significance of the 3.7-kb deletion in Black individuals?
It is seen in approximately 30% of Black people and is a common form of Alpha Thalassemia.
What happens to the a2 production in individuals with a 1 gene deletion?
The a2:a1 ratio becomes 1:1, and loss of a2 results in compensatory increase in a1 production.
What is the typical laboratory finding in CBC for Alpha Thalassemia?
Low MCV, low MCH, and high RDW.
What is the relationship between excess beta chains and RBCs in Hemoglobin H disease?
Excess beta chains accumulate inside RBCs, damaging the membrane and impairing oxygen transport.
What is the clinical significance of identifying Alpha Thalassemia carriers?
It is important for genetic counseling and understanding potential health impacts.
What is the expected laboratory finding for a silent carrier of Alpha Thalassemia?
Normal findings with no anemia.
What is the result of having two gene cis deletions in alpha globin genes?
Very severe Hemoglobin H disease.
What are the clinical features of HbH disease with a 2-gene deletion?
More severe anemia, hemolysis, splenomegaly, and increased need for transfusions.
What is Hemoglobin Barts and how is it formed?
It is formed by the deletion of all 4 alpha chains, leading to fetal γ chains associating in groups of 4 (γ4).
What percentage of newborns with 1 or 2 gene deletions may show Hb Barts on screening?
Usually <5% with 1 gene deletion; 5-10% with 2 gene deletion; 15-20% with Hb H disease.
What is the significance of Hb Barts on HPLC testing?
It is used to rule out alpha thalassemia by DNA testing.
What types of testing are commonly used for alpha globin DNA analysis?
Common mutations testing, sequencing for point mutations and variants, and dosage testing for rare deletions and duplications.
What is the most common cause of alpha thalassemia?
85-90% of people with alpha thalassemia have deletions.
What should a healthcare provider do first to rule out alpha thalassemia in a patient?
Order testing for deletions before sequencing.
What is the origin of the term 'Thalassemia'?
It is derived from the Greek word 'Thalassa', meaning sea.
What are the two types of mutations that cause beta thalassemia?
Point mutations and large deletions.
What are the clinical features of beta-thalassemia minor (trait)?
Mild anemia, decreased MCV and MCH, increased Hb A2 (4%-6%), and normal or slightly increased Hb F (2%-5%).
What characterizes beta-thalassemia intermedia?
Milder symptoms than beta thalassemia major but more severe than just trait, due to a more normal alpha/beta synthesis ratio.
What are the clinical features of beta-thalassemia major (Cooley's Anemia)?
Severe microcytic hemolytic anemia, massive destruction of nucleated RBCs, requires transfusions every 4-8 weeks, and hepatosplenomegaly.
What complications are associated with beta-thalassemia major?
Cholelithiasis (gallstones), susceptibility to infections, and delayed growth and maturation.
What happens to the bone marrow in beta-thalassemia major?
There is massive destruction of nucleated RBCs and expanded bone marrow cavities.
What is the effect of mutant alpha2 genes in alpha thalassemia?
They likely retain normal transcription but produce no protein product, interfering with normal alpha1 gene transcription.
What is the role of the Locus Control Region (LCR) in beta globin gene expression?
It regulates the expression of globin genes including HBE, HBG1, HBG2, HBD, and HBB.
How does beta-thalassemia major affect growth and maturation in patients?
It leads to delayed growth and maturation due to severe anemia and related complications.
What is the typical treatment requirement for patients with beta-thalassemia major?
They require regular blood transfusions.
What are the common symptoms of hepatosplenomegaly in beta-thalassemia major?
Enlarged liver and spleen due to extramedullary hematopoiesis.
What is the significance of increased Hb A2 levels in beta-thalassemia minor?
It indicates a compensatory response to the decreased production of normal beta globin.
What characterizes Beta-thalassemia intermedia?
Patients exhibit anemia and hepatosplenomegaly but do not present the full spectrum of symptoms found in Beta-thalassemia major.
What is a common genetic cause of Beta-thalassemia?
The vast majority of Beta-thalassemia cases are due to point mutations, with 95% of carriers having a point mutation and 5% having large deletions.
What is the first test to order to rule out Beta-thalassemia?
Beta globin sequencing is the best initial test as it identifies point mutations and variants.
What does Beta globin dosage testing identify?
It identifies large deletions and duplications, such as the common 619 bp deletion found in Asian Indian populations.
What is the significance of Hereditary Persistence of Fetal Hemoglobin (HPFH) in Beta-thalassemia?
HPFH is usually due to the deletion of the delta and beta globin genes and is very common in African Americans.
How does high HbF relate to Beta-thalassemia testing?
If HbF is very high, there is a high chance of delta-beta deletion, indicating that dosage testing should be performed instead of sequencing.
What is the clinical utility of knowing a patient's ethnicity when testing for Beta-thalassemia?
Ethnicity may influence the likelihood of specific mutations, such as the 619 bp deletion in Punjabis.
What is the main goal of verification in Beta-thalassemia testing?
To ensure that the healthcare provider receives the necessary information to care for the patient and to avoid unnecessary testing.
What does a negative test result imply in the context of Beta-thalassemia?
A negative result should be powerful enough to save the patient time and money by preventing additional testing.
What are the clinical features of Beta-thalassemia major?
Patients typically present with severe anemia, requiring regular blood transfusions and exhibiting various complications.
What does a hemogram indicating elevated A2 levels suggest?
It may suggest a beta plus mutation, but further investigation is needed to determine if the patient is homozygous or compound heterozygous.
What mutation is indicated by a result of c.-79A>G?
This mutation is a promoter mutation that typically confers a very high A2 level.
What does a lower than expected A2 level indicate in a patient with a c.-79A>G mutation?
It suggests that the patient may have one copy of the mutation and a deletion on the other chromosome, indicating a compound heterozygous state.