20. Homeostasis: Regulation of Intracellular Calcium
1/11
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
12 Terms
How does calcium function as an ON/OFF switch in muscle and other cells?
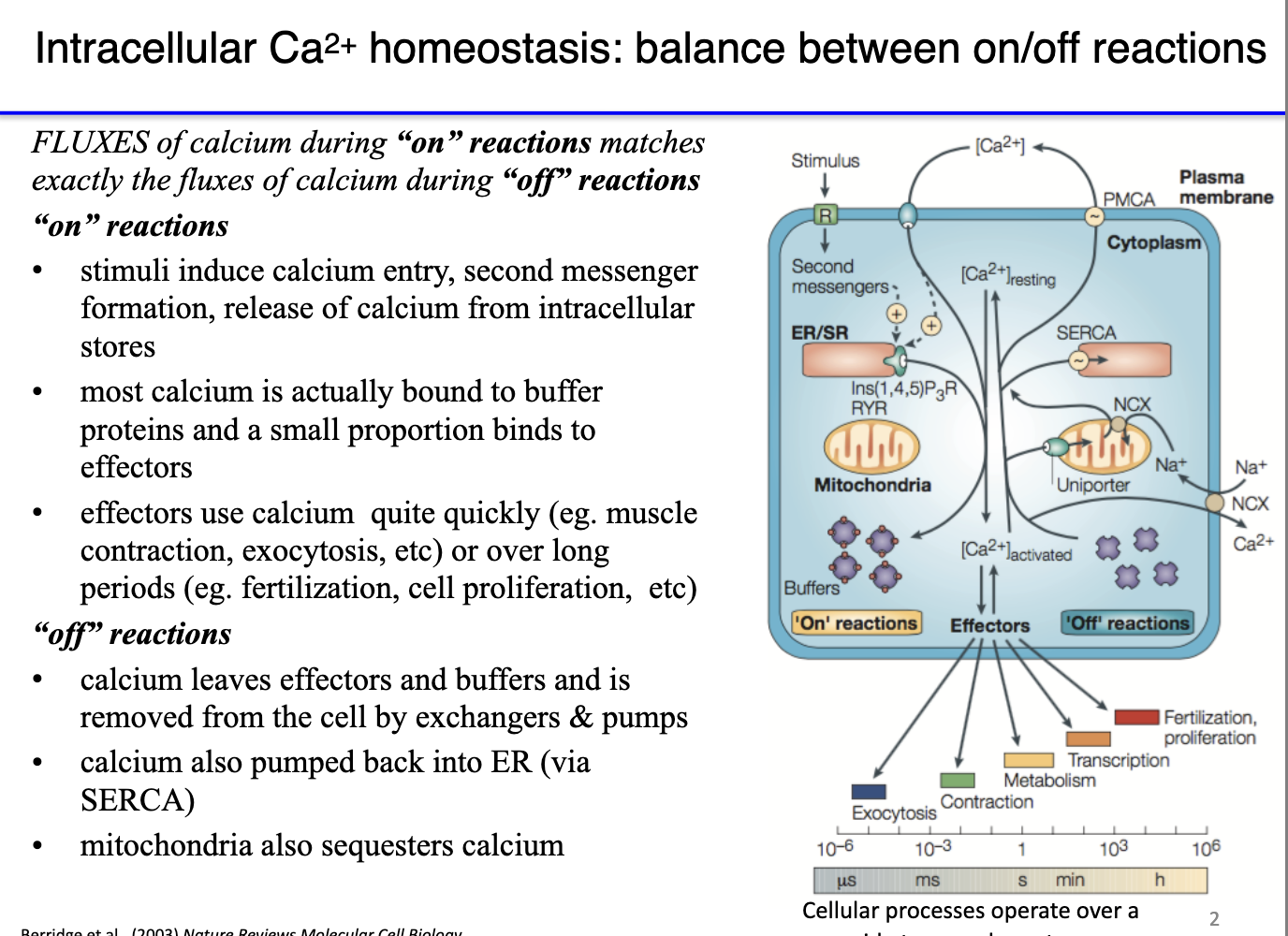
ON reaction: Ca²⁺ floods into cytoplasm → binds effector proteins (e.g., troponin) → exposes myosin binding sites → triggers contraction or other Ca²⁺-activated pathways.
OFF reaction: Ca²⁺ must be rapidly removed so it dissociates from effectors:
PMCA pumps Ca²⁺ out of the cell (ATP-dependent).
Na⁺/Ca²⁺ exchanger exports Ca²⁺ via secondary active transport.
SERCA returns Ca²⁺ to the SR.
Ca²⁺ is not degraded, only sequestered into compartments where it can’t activate proteins.
Muscle function depends on Ca²⁺ cycling: brief cytoplasmic spikes (ON) followed by rapid clearance (OFF).
This fast ON/OFF cycling enables repeated, coordinated contractions across whole muscles.
How does the mitochondrial calcium uniporter (MCU) complex regulate Ca²⁺ entry into the matrix?
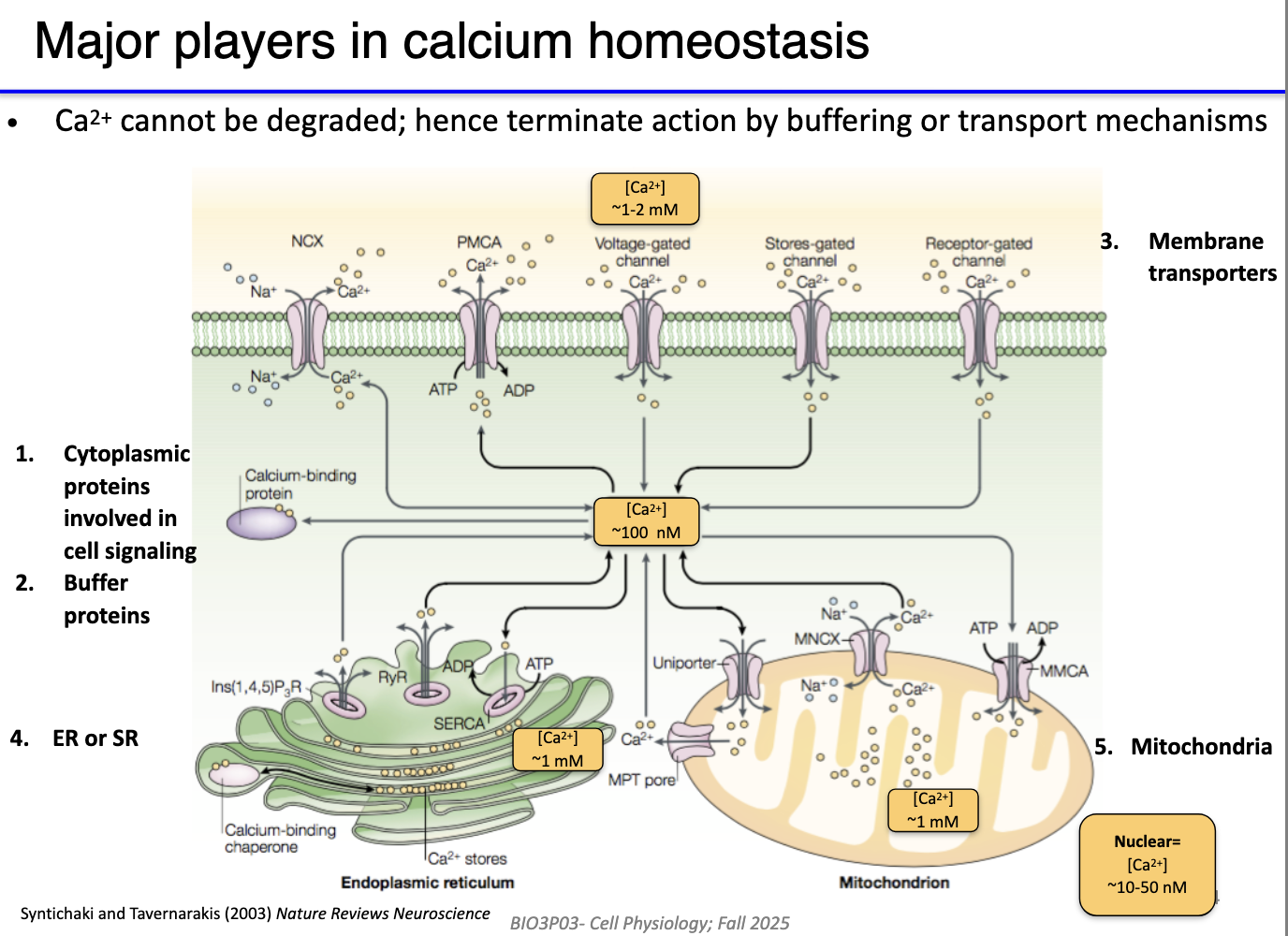
Resting cytoplasmic Ca²⁺ ~100 nM (very low).
High Ca²⁺ stores:
Extracellular fluid: 1–2 mM
SR/ER: very high
Mitochondria: high (similar to SR)
Low Ca²⁺ areas: cytoplasm, nucleus.
Key Ca²⁺ regulators:
VG Ca²⁺ channels → Ca²⁺ influx.
PMCA → pumps Ca²⁺ out (ATP-dependent).
Na⁺/Ca²⁺ exchanger → secondary active export.
IP₃R & RyR → Ca²⁺ release from SR/ER.
SERCA → returns Ca²⁺ to SR/ER.
Homeostasis must support rapid spikes (ON) and rapid clearance (OFF) while maintaining stores for future signaling.
What roles do calmodulin and Ca²⁺-buffering proteins play in calcium signaling?
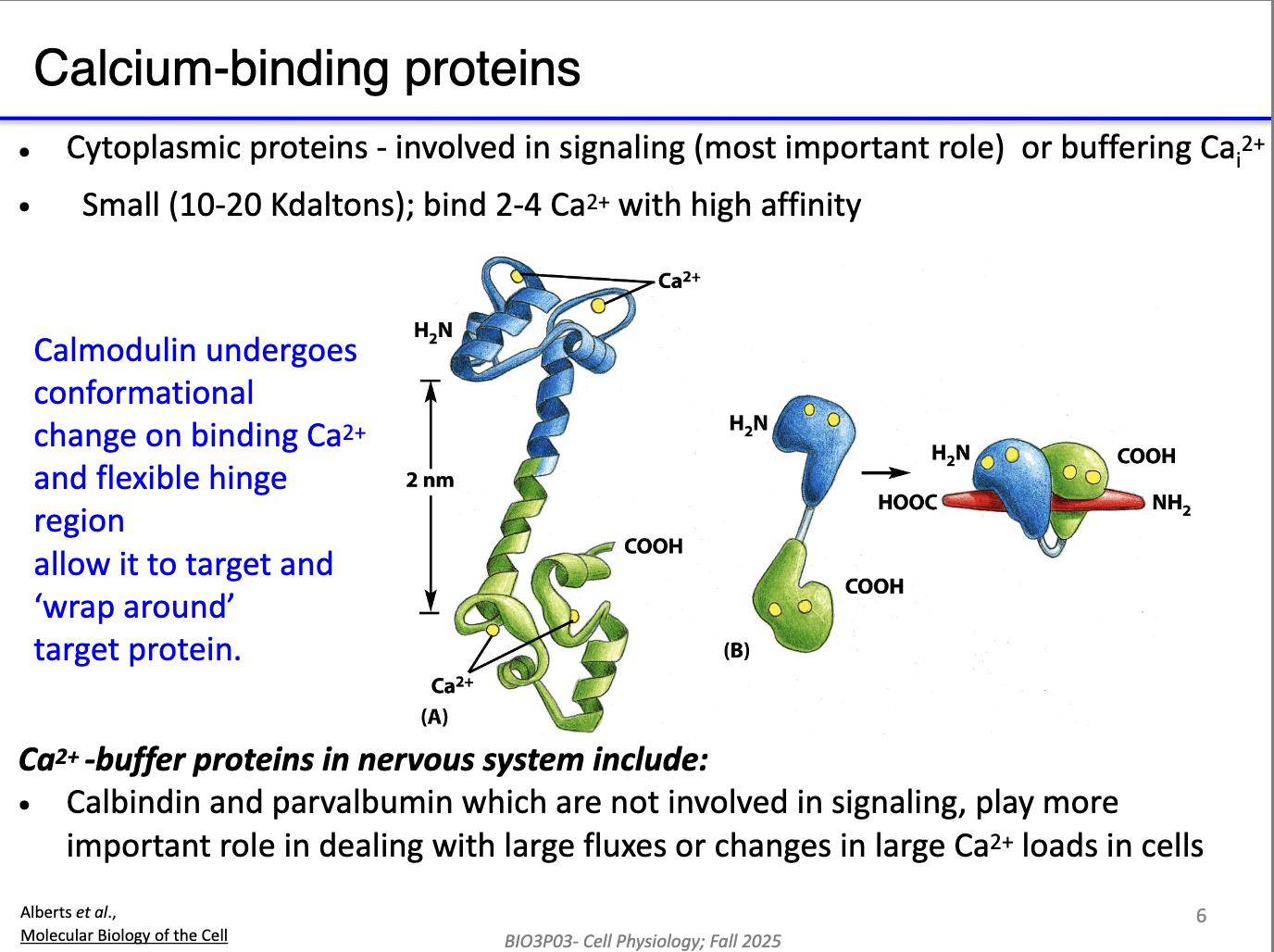
Calmodulin:
Monomer with two globular domains + hinge.
Ca²⁺ binding → conformational change → wraps around target proteins → alters the protein activity.
Ca²⁺ buffer proteins (e.g., calbindin, parvalbumin):
Bind Ca²⁺ quickly to reduce free Ca²⁺.
Temporarily block Ca²⁺ from activating effectors.
Speed the OFF reaction by limiting Ca²⁺ availability before pumps fully clear it.
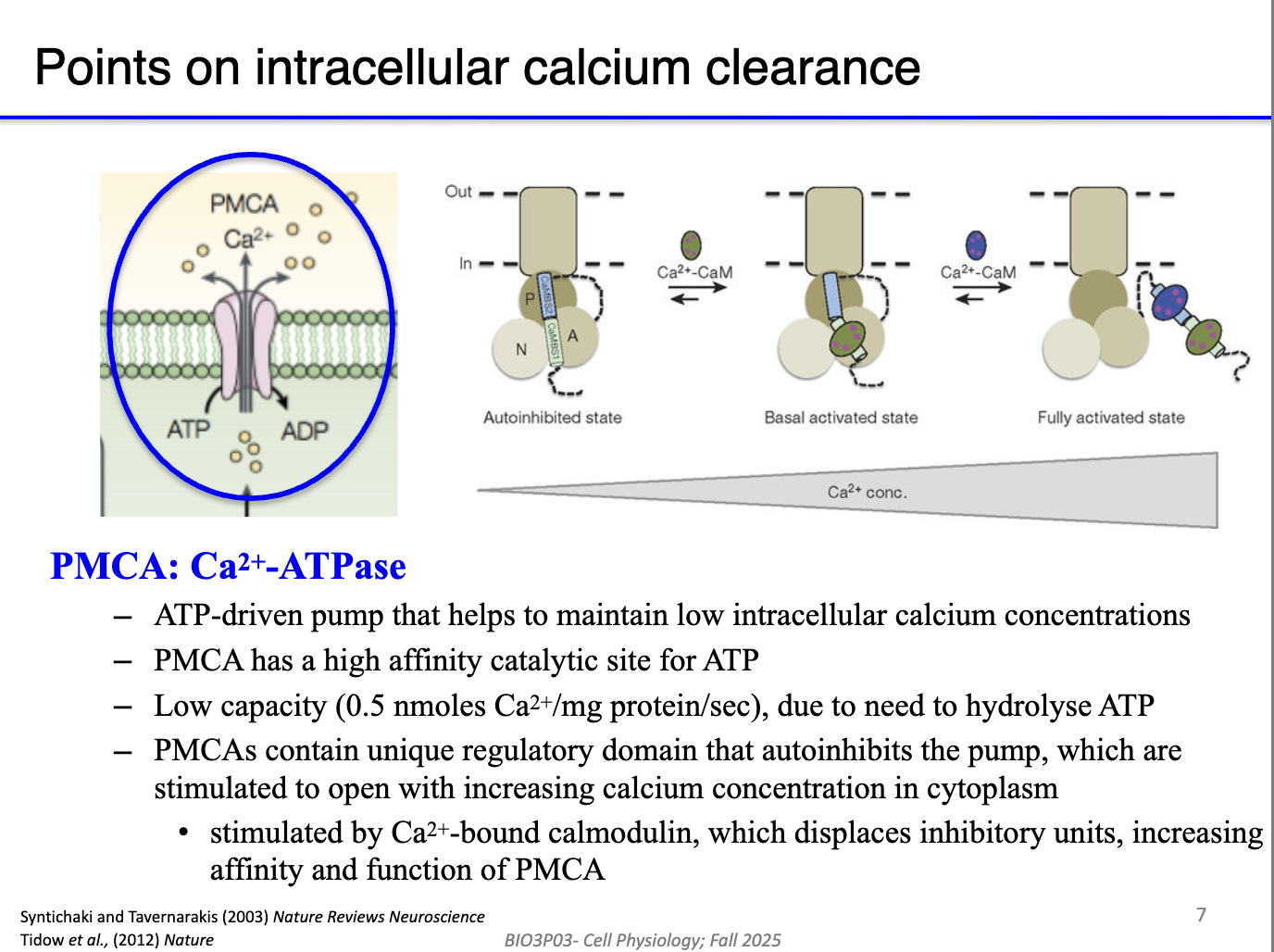
How does calmodulin regulate PMCA to control Ca²⁺ clearance?
PMCA pumps Ca²⁺ out of the cell but is autoinhibited at rest to avoid wasting ATP.
High cytosolic Ca²⁺ → Ca²⁺ binds calmodulin → Ca²⁺/CaM complex binds PMCA’s inhibitory domain.
This removes autoinhibition → activates PMCA → rapid Ca²⁺ export.
As Ca²⁺ falls, Ca²⁺ unbinds calmodulin → PMCA becomes autoinhibited again.
Creates a feedback system: PMCA turns on only when Ca²⁺ is high.
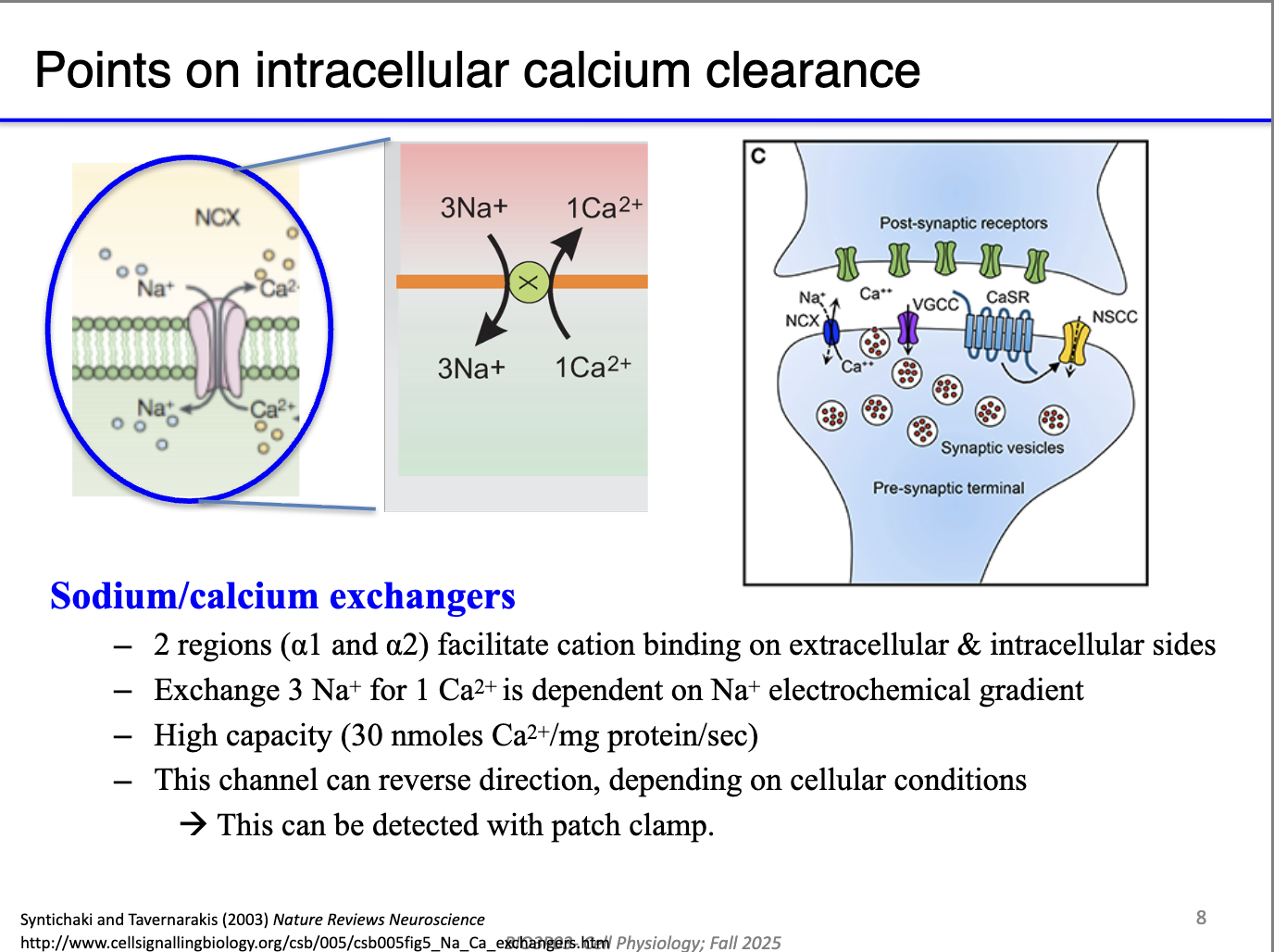
How does the Na⁺/Ca²⁺ exchanger contribute to Ca²⁺ clearance?
Secondary active transport: uses Na⁺ gradient (maintained by Na⁺/K⁺-ATPase) to export Ca²⁺.
Fast, always running in the background—no calmodulin required.
Critical in presynaptic terminals, where Ca²⁺ must be cleared quickly to stop continuous vesicle fusion.
Prevents excess transmitter release by removing Ca²⁺ after each action potential.
Why does Ca²⁺ flow into mitochondria, and what drives it?
Mitochondria have two membranes; Ca²⁺ passes outer membrane via VDAC channels.
Inner membrane Ca²⁺ entry is driven by the strongly negative mitochondrial matrix (~–200 mV).
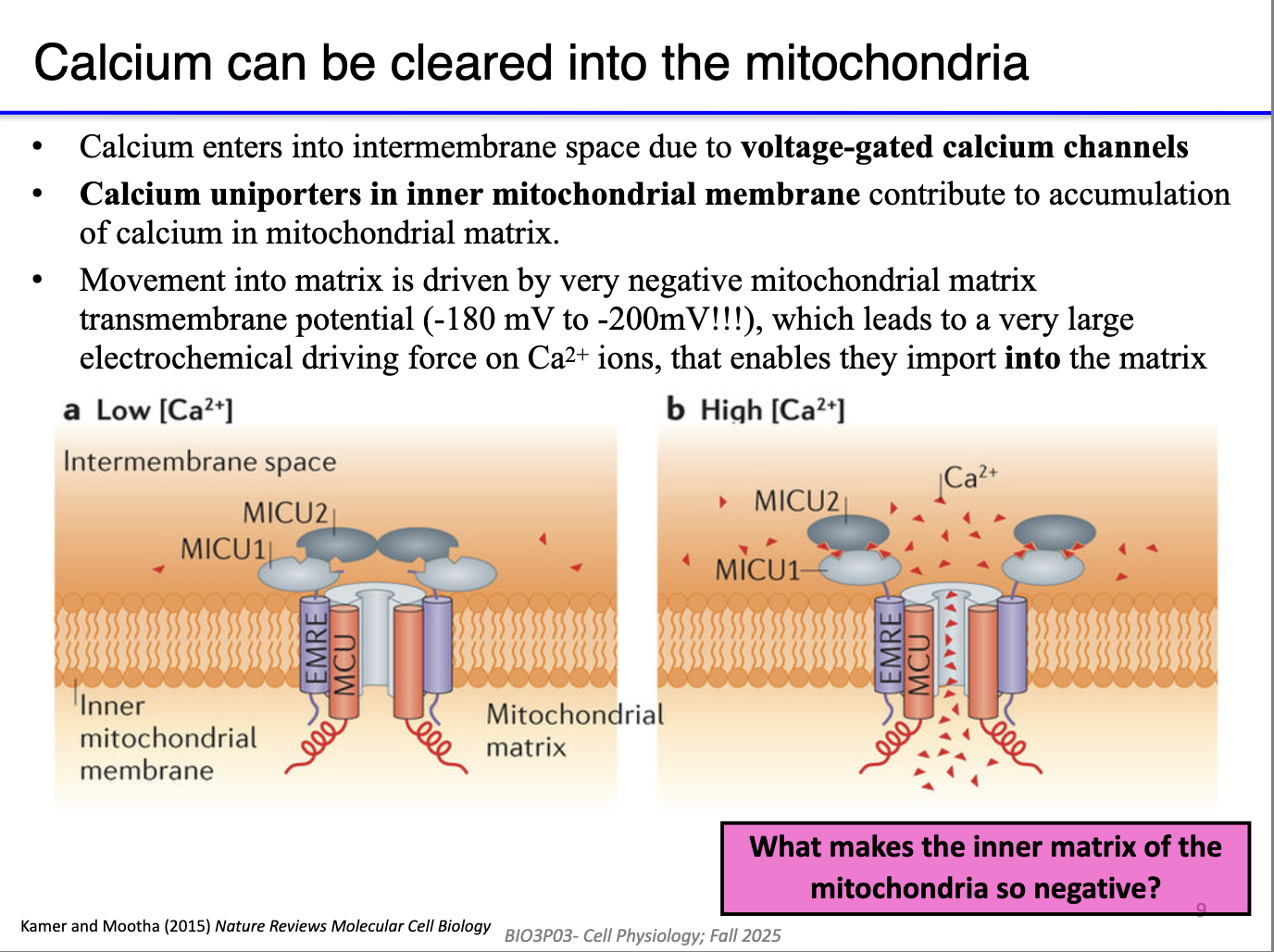
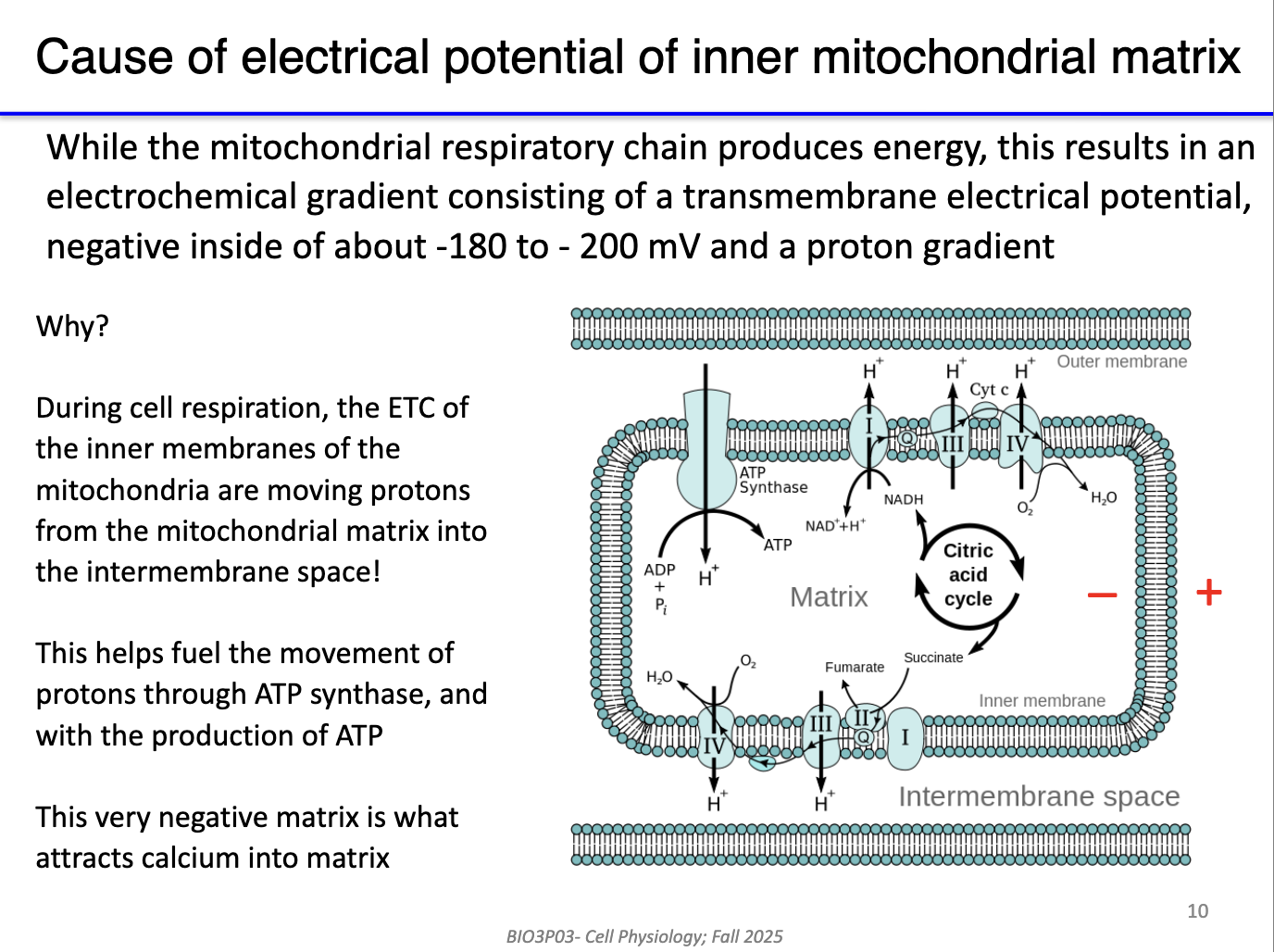
What makes the inner mitochondrial matrix so negative?
This negativity results from the electron transport chain pumping protons into the intermembrane space.
No pumps required: Ca²⁺ moves via channels because of the large electrical gradient.
Mitochondria act as a secondary Ca²⁺ reservoir and use Ca²⁺ to stimulate metabolic enzymes.
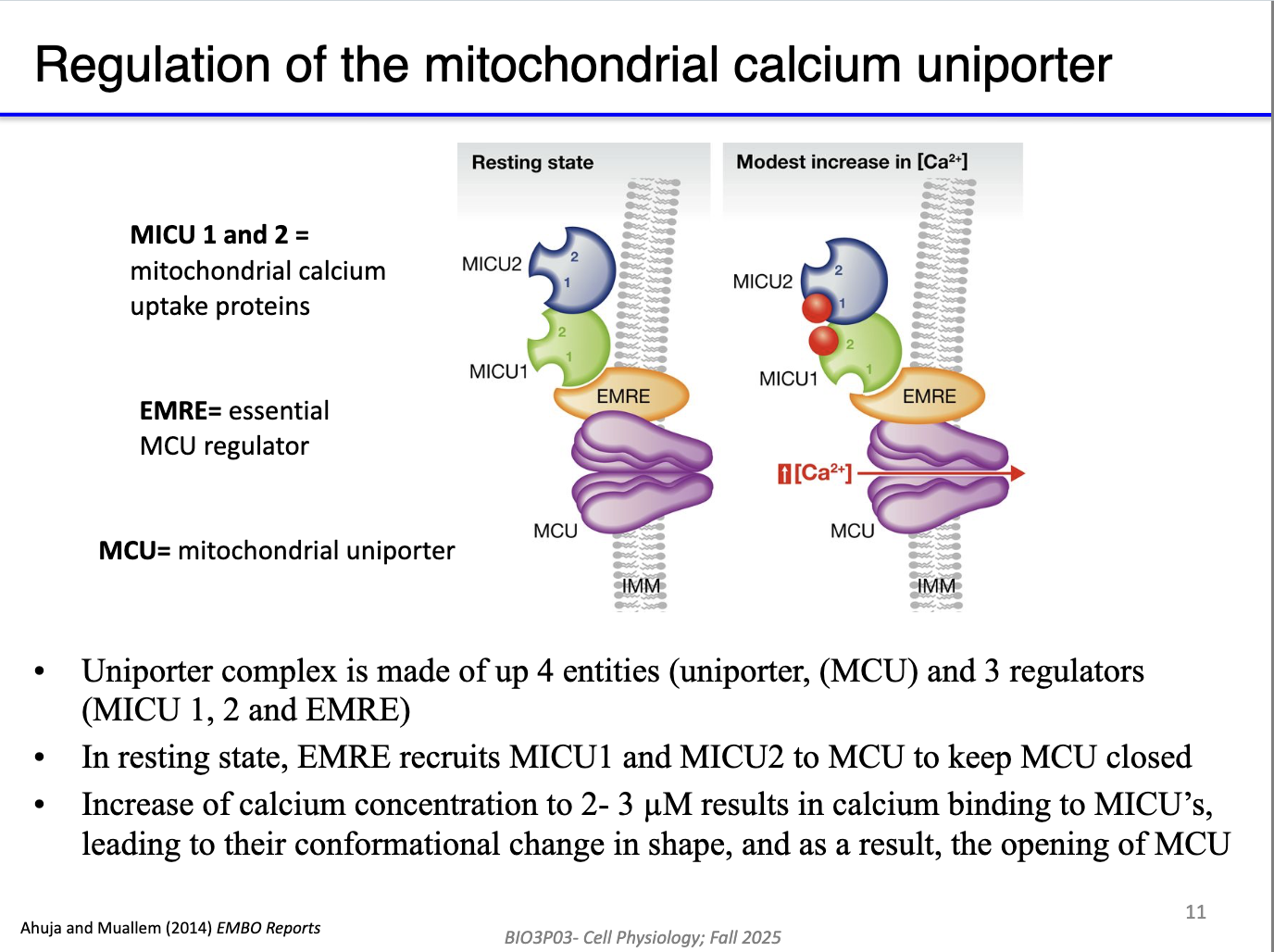
How does the mitochondrial calcium uniporter (MCU) complex regulate Ca²⁺ entry into the matrix?
MCU (inner membrane channel) allows Ca²⁺ entry when open.
EMRE (MCU regulator) organizes the complex.
MICU1/2 are Ca²⁺-binding gatekeepers:
Low Ca²⁺ → MCU closed.
High Ca²⁺ → MICU1/2 bind Ca²⁺ → conformational change → MCU opens.
Allows controlled Ca²⁺ entry despite huge electrical driving force.
Mitochondria store Ca²⁺ and also use it to stimulate the Krebs cycle & ETC.
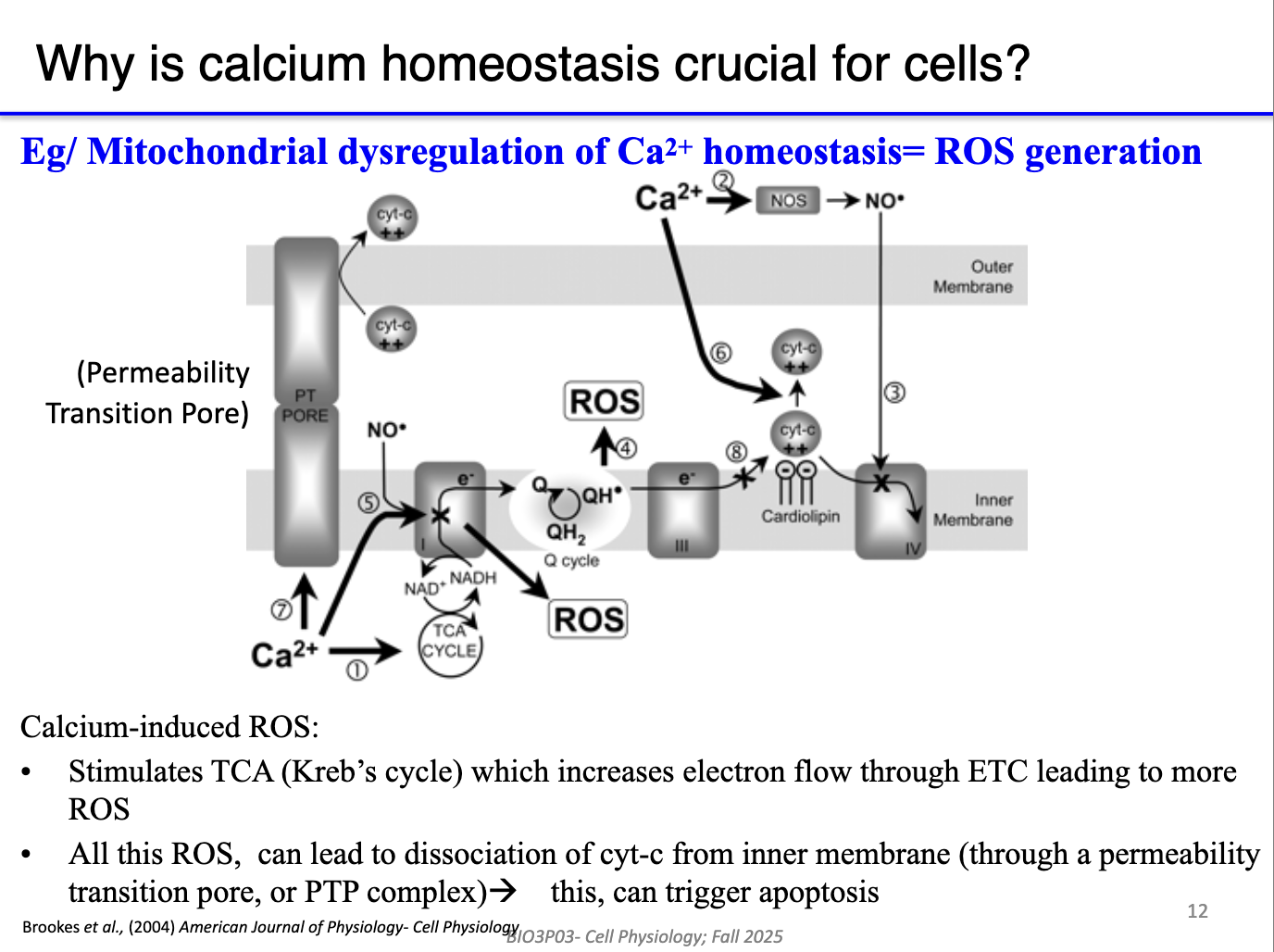
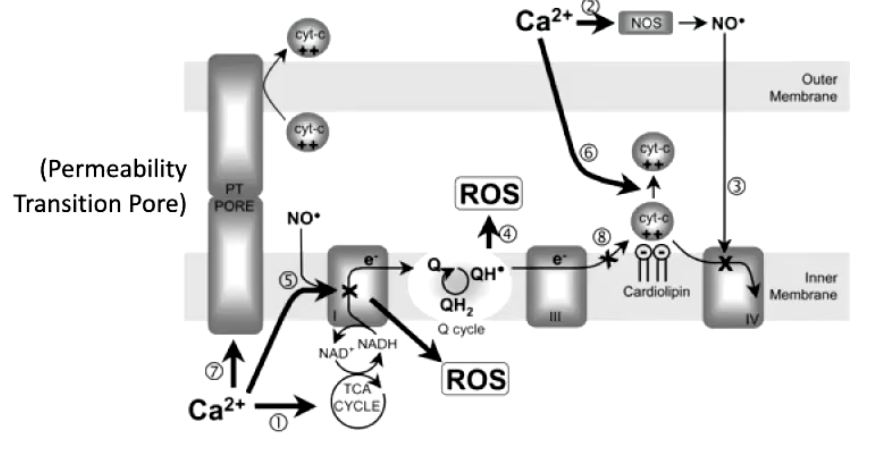
What happens when mitochondria take up too much Ca²⁺?
Excess matrix Ca²⁺ → overactivation of Krebs cycle + ETC → excessive proton pumping.
Leads to ROS (reactive oxygen species) production at the electron transport system.
ROS damage:
DNA, proteins, lipids (especially phospholipid inner membrane).
Causes cytochrome c leakage, triggering apoptosis.
Mitochondrial Ca²⁺ overload = major trigger for cell death.
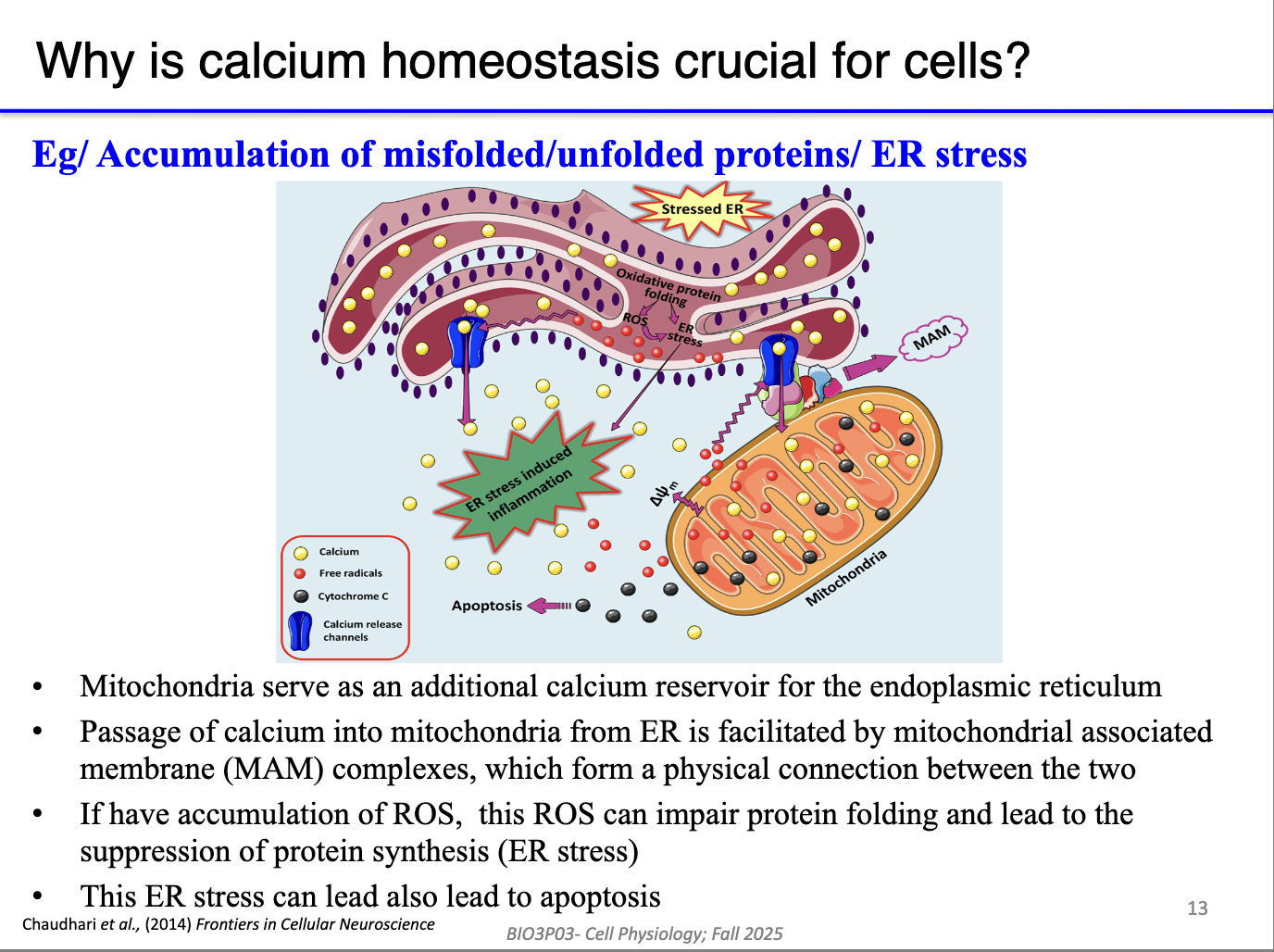
How do mitochondria and ER/SR share Ca²⁺, and why does it matter?
Linked via MAMs (mitochondria-associated membranes) → direct Ca²⁺ and stress signal transfer.
Mitochondria act as a reserve reservoir:
Can return Ca²⁺ to ER/SR when stores run low.
However, ROS or Ca²⁺ overload in mitochondria can spread stress to ER/SR → ER stress → apoptosis.
MAMs allow both beneficial Ca²⁺ sharing and propagation of damage.
Why does reperfusion after ischemia cause dangerous mitochondrial Ca²⁺ overload?
During ischemia/hypoxia (low O₂):
ETC stops → no proton gradient.
Ca²⁺ pumps (PMCA, SERCA) fail due to low ATP.
Na⁺/Ca²⁺ exchanger fails because it relies on the gradients set up by Na+/K+ pump, which uses ATP.
Ca²⁺ builds up in cytoplasm but does not enter mitochondria (no negative matrix).
During reperfusion (O₂ returns):
ETC restarts instantly → matrix becomes highly negative before Ca²⁺ pumps recover.
Massive Ca²⁺ rushes into mitochondria → ROS burst → membrane damage → cytochrome c release → apoptosis.
This is why reperfusion injury is worse than the ischemia itself.
How do mitochondria safely store Ca²⁺ without triggering damage?
Mitochondria import inorganic phosphate (Pi).
High Ca²⁺ + Pi → form calcium phosphate precipitates.
Precipitated Ca²⁺ = inactive, preventing:
Excess Krebs cycle stimulation
Excess ETC flux
ROS generation
When ER/SR needs Ca²⁺, free Ca²⁺ diffuses out → due to low Ca2+ in mitochondria, precipitates dissolve → replenish Ca²⁺ stores.
Buffering prevents mitochondrial Ca²⁺ overload except under extreme stress (e.g., ischemia/reperfusion).
