Biochem Quiz 8
1/40
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
41 Terms
Intracellular Proteolysis
removes mis folded, old, or damaged proteins
Supplies essential amino acids when diet is not enough
Controls cell-cycle transitions
Digestion of Dietary Proteins Provides
Nutritionally essential and nutritionally nonessential amino acids
De Novo Synthesis
Provides nutritionally nonessential amino acids needed for synthesis
Adjusts amino acid pools
Adjusts energy metabolism
Needed to make nucleotides, genes, hormones
9 Essential Amino Acids
Tryptophan
Phenylalanine
Leucine
Isoleucine
Lysine
Histidine
Threonine
Methionine
Valine
3 Conditionally Essential Amino Acids
Arginine
Tyrosine
Cysteine
Proteostasis
Protein homeostasis
Maintained by protein turnover
3 Protein Degradation Processes
Lysosome
Proteasome
Autophagic Path
Overview: Ubiquitin Proteasome System
Selective degradation
Highly selective
In cytosol and nucleus
ATP dependent
Autophagy Overview
Bulk degradation
Initiated in cytosol
Requires energy
Overview Lysosomal Degradation
Degradation of endocytosed protein, membrane proteins
Less selective
At lysosome
Contributes to amino acid pool
Ubiquitination
ATP → PPi + AMP
Ub loaded to E1 (initiating enzyme)
Ub to E2 (conjugating enzyme)
Ub to E3 (protein ligase enzyme)
Polyubiquitination possible
Proteasome degrades
Zymogens
enzymes released in this form
Prevents self digestion
Only activate in certain conditions
Activation: proteolytic cleavage
Stomach (Initiation of Digestion)
Pepsinogen → Pepsin
Low pH
Activation is auto catalytic
Initial protein cleavage
Small Intestine (Amplification of Degradation))
Trypsinogen → Trypsin
Activated in intestinal lumen
Trypsin activates other proteases
Absorption
in small intestine across enterocytes
via transporters
in cell peptides → amino acids
then amino acids → portal circulation → liver
Amino Acid Transport
secondary active transport
gradient via Na/K pump
When are amino acids oxidized?
when during protein turnover they are released but not needed for synthesis
when ingested amino acids exceeded needed amount
when cellular proteins are used as fuel bc carbs are not available
Pyruvate is the keto-acid of ________ .
Alanine

Oxaloacetate is the keto-acid of ________ .
Aspartate
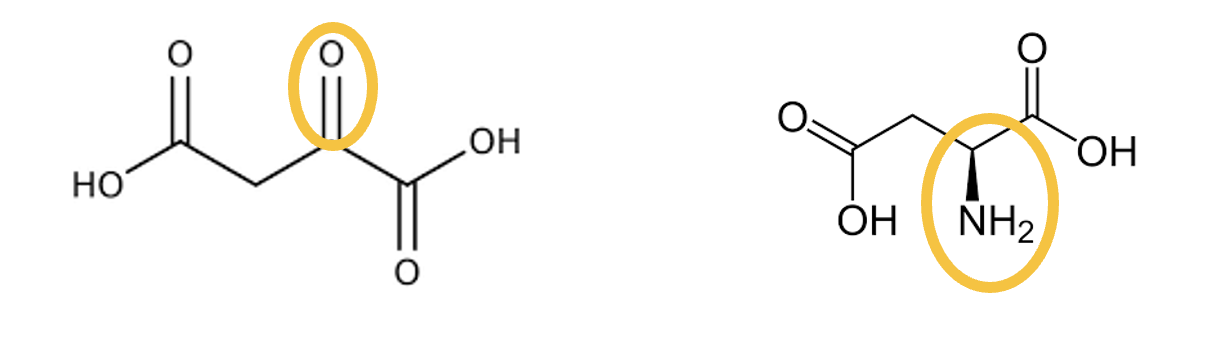
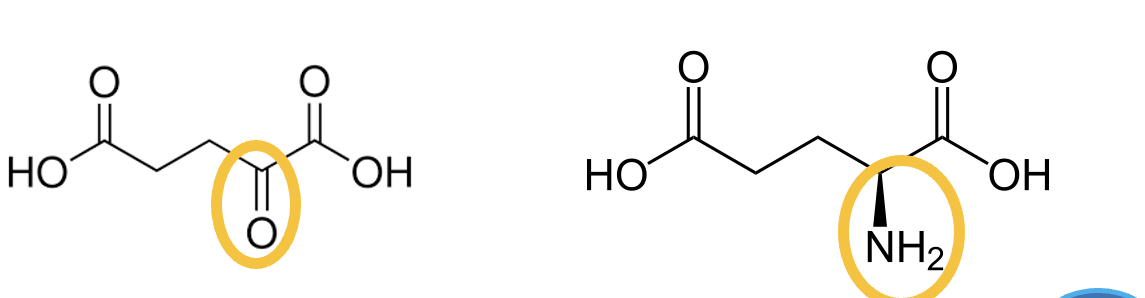
Alpha-Ketoglutarate is the alpha-keto-acid of ________ .
Glutamate
Transamination Overview
Removal of amino group from amino acids
Needed step in amino acid catabolism
In cytosol
Amino group transferred to alpha carbon of alpha keto acid
Leaves behind alpha-keto acid analog of amino acid
Enzyme Type in Transamination (2)
Aminotransferases
Transaminases
PLP
Needed in transamination reactions
In first step amino group transferred onto PLP making it PMP
Derivative of Vitamin B6
Prevents release of amino group to environment
Alanine Transferase (ALT)
Reversible
Reactants: Alanine + a-KG
Products: Pyruvate + Glutamate
Aspartate Transferase (AST)
Reversible
Reactants: Aspartate + a-KG
Products: OAA + Glutamate
Proline and Hydroxyproline
Secondary amines
Cannot undergo transamination reactions
Lysine and Threonine
Do not undergo transamination
Form toxic nonmetabolites if they do
Glutamate Dehydrogenase (GDH) Overview
Major route for oxidative deamination
Can also do reductive amination
Regenerates amino acceptor (a-ketoglutarate)
Provides ammonia (NH4)
In mitochondrial matrix
2 step process
Oxidative Deamination
Reactant: Glutamate
Enzyme: GDH
NAD+ reduced to NADH
H2O added in second step
Products: a-KG and NH4+
Reductive Amination
Reactants: a-KG and NH4+
Water produced
NADPH oxidized to NADP+
Product: Glutamate
True or False: No net deamination in transaminase reactions because a-kg becomes aminated.
true
Allosteric control of GHD dependes on?
Cellular energy state
GDH Regulation: HIgh ATP, GTP, NADH
GDH inhibited
Protein Synthesis initiated instead
GDH Regulation: High ADP, GDP, Free Amino Acids
GDH activated
Deamination initiated
Glutaminase
Other deamination route
Gultamine → Glutamate
Ammonia formed is consumed by urea cycle
Widely distributed in body
Asparaginase
Other deamination route
Asparagine → Aspartate
Reaction & Role of Glucose-Alanine Cycle
Toxic ammonia transported to liver as alanine
Alanine formed in muscle via transamination of pyruvate
In liver alanine reconverted to pyruvate
Pyruvate used to create glycolysis via gluconeogenesis
Malate-Aspartate Shuttle & How it Supports Nitrogen Disposal
Transfers reducing equivalents across mitochondrial membranes
Regeneration of OAA
Can accept an amino group to form aspartate
Aspartate to step 2 of urea cycle
Maintains availability of aspartate
PLP deficiency ______ Transamination. Why?
Decreases
Required coenzyme for aminotransferases
PLP deficiency _____ Urea Cycle flux. Why?
Decreases
Fewer amino groups reach glutamate
Decrease in NH4+ production via GDH
PLP deficiency ____ TCA Cycle flux. Why?
Decreases
a-KG, OAA, pyruvate production from amino acids decrease