Pharmacokinetic Studies and Measurements of Drug Concentration (slide 6-36)
1/61
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
62 Terms
Pharmacokinetic (PK) studies
are used to find out therapeutic doses with an adequate profile of drug concentration versus time.
Approaches of Pharmacokinetic studies
Experimental Approach (blood conc.)
Theoretical Approach
Experimental Approach
Involves the development of biologic sampling techniques, analytical methods for the measurement of drugs and metabolites, and procedures that facilitate data collection and manipulation.
Based on assumptions only
Pharmacokinetic Model
Mathematical representation of drug movement over time
Pharmacokinetic
Mathematical representation of ADME
Theoretical Approach
Involves the development of pharmacokinetic models that predict drug disposition after drug administration
Statistical methods are used for pharmacokinetic parameter estimation and data interpretation ultimately for the purpose of designing and predicting optimal dosing regimens for individuals or groups of patient.
Base on given data only
Measurements of Drug Concentration
Because drug concentrations are an important element in determining individual or population pharmacokinetics, drug concentrations are measured in biologic samples, such as milk, saliva, plasma, and urine.
Sensitive, accurate, and precise analytical methods are available for the direct measurement of drugs in biologic matrices.
Direct measurement
Measurements will vary depending on the sample
There is a relationship in drug dose and pharmacodynamic response
Two types of methods of (measurement)
Invasive method
Extensive method
Invasive method
methods that needs penetration inside the body (ex. Vaccine)
Extensive method
methods that don’t need penetration inside the body
Blood flow
are different in different areas so we can say that blood concentrations are different also
Drug concentration in tissue biopsies
may not reflect drug concentration in all parts of the tissue from which the biopsy material was removed
Drug concentration in urine
Indirect method to ascertain the bioavailability of a drug.
Rate and extent of drug in the urine may be related/reflect to the rate and extent of
Drug concentration in feces
reflect the concentration of drug that has not been absorbed after an oral dose.
Drugs that are absorbed via liver is excreted via bile or feces
Drug concentration in saliva
tend to approximate free drug rather than total plasma drug concentration.
Highly lipophilic drugs only drugs that can pass through
Plasma Isolation
Collect blood in anticoagulated tubes (EDTA, citrate, heparin)
Centrifuge blood sx
Pipette plasma
Apply to cells (1-100% v/v)
Serum Isolation
Collect blood in tube (no anticoagulant)
Incubate sx until blood fractionation
Pipette serum
Apply to cells (1-100% v/v)
Bound drug
drug is binded in the albumin
How do we do Pharmacokinetic studies
Calculate how fast the drug appears in the blood (To determine the rate and extent absorption)
Measure how much of the drug is in the blood (Volume of distribution)
Calculate how fast the drug disappears. (Excretion)
Pharmacokinetics
is the effect of body on the drugs.
Physiology
determines Pharmacokinetics.
Key Pharmacokinetic Parameters
Dose
Bioavailability
Volume of Distribution
Absorption/Elimination Rate Constant
Clearance
Dose
The amount of drug administered to achieve a desired effect.
Bioavailability
The fraction of the administered dose that reaches the systemic circulation in active form.
Volume of Distribution (Vd)
The theoretical volume that would contain the drug at the same concentration as in the blood.
Absorption/Elimination Rate Constant (Ka/Ke)
The rate at which a drug is absorbed into or removed from the body.
Clearance (Cl)
The volume of plasma cleared of drug per unit time, reflecting the body’s ability to eliminate the drug.
Kinetic Homogeneity
Efficacy of a drug product depends on the drug concentration at the targeted site of a human body after administrating such drug product.
Drug concentration
is derived by collecting a blood sample at any time after drug administration and measuring the amount of a drug in a given volume of blood plasma of the sample.
Characterize the therapeutic effect
A profile of plasma concentration versus time can be used to ___________ of a drug for a specific dosage or formulation
At time 0
which is the moment before the drug is administered or just as it is given, there is typically no drug yet in the systemic circulation.
IV
Max conc. Reached at zero time
Effects starts at zero time (There’s onset of action already)
No absorption = Precise Dose
EV
Max conc. reached after a certain TIME
Has absorption phase
Effects starts after reaching MEC
Elimination
Both IV and EV have an _______ phase
Plasma Level vs. Time Curve (Single dose)
See image below
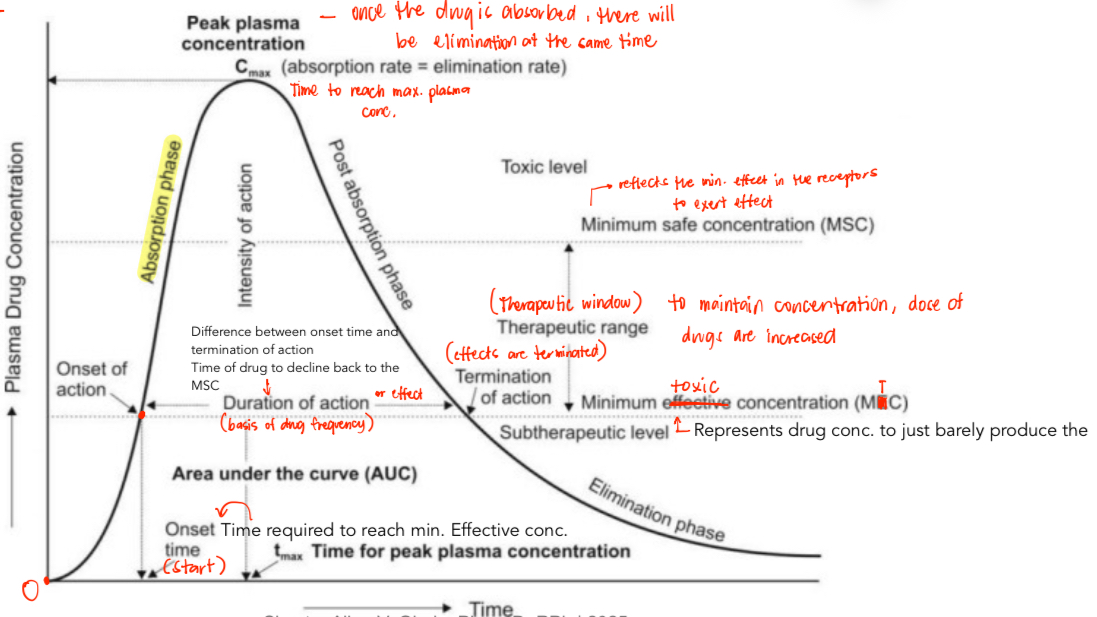
Plasma Level vs. Time Curve
See image below
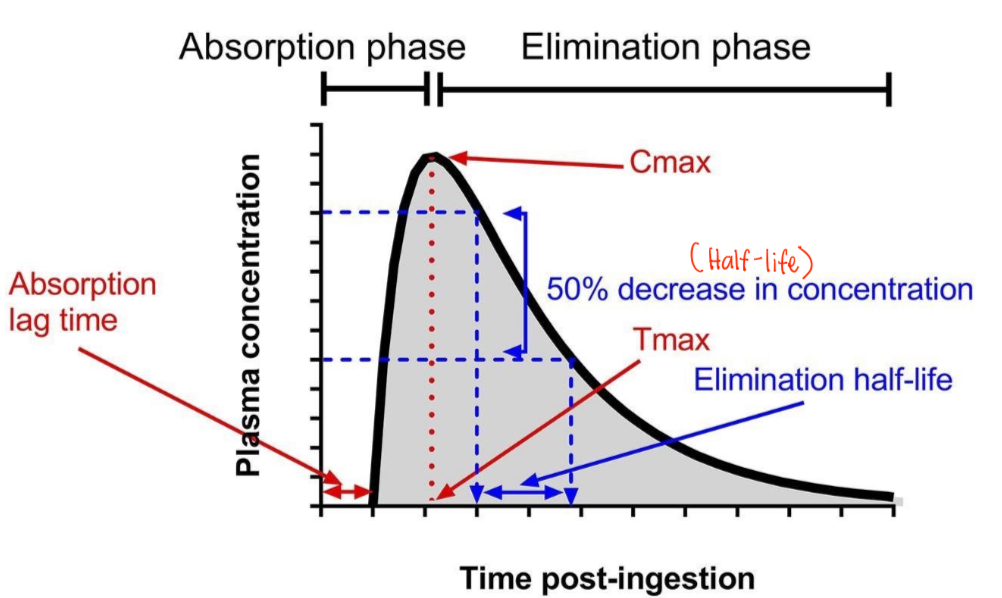
Cmax
Measures INTENSITY of effects
Increase Cmax = increase response
AUC
Amount of drug in the blood stream/time = EXTENT of absorption (Basic principle of kinetic homogeneity)
Increase amount = increase effects
Tmax
Measures the rate of absorption
Increase Tmax = decrease rate of absorption
Pharmacokinetic Parameters Used to Assess Bioavailability
Tmax – Rate
Cmax – Rate and Extent (Intensity of Response)
AUC – Extent
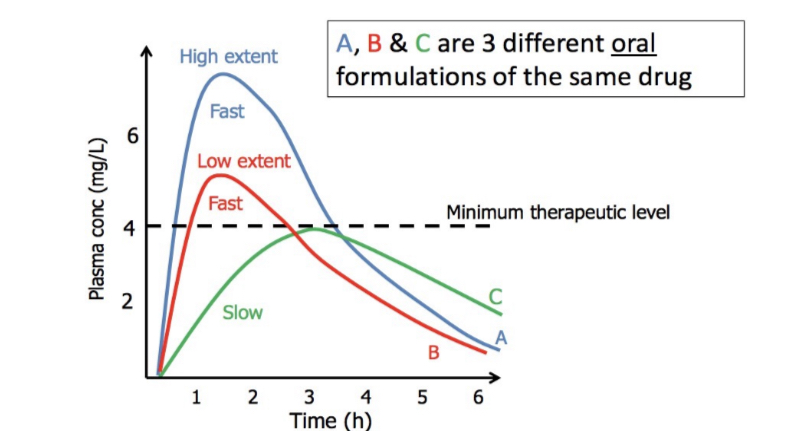
Pharmacokinetic Parameters Used to Assess Bioavailability
A, B, & C are 3 different oral formulations of the same drug
Laws of Exponents
See image below

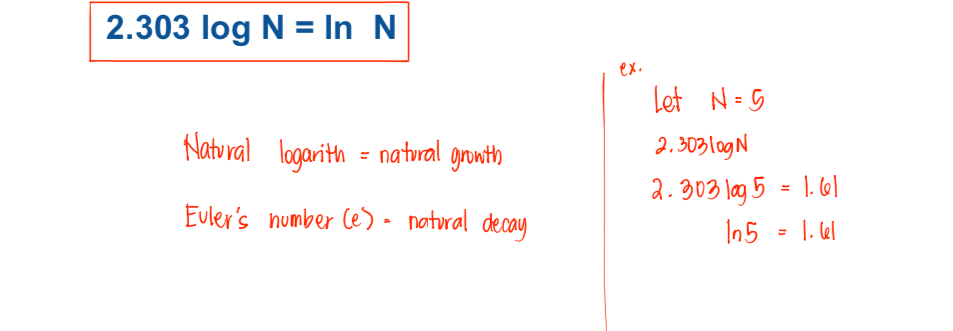
Logarithm
the _________ of a positive number N to a given base b is the exponent x to which the base must be raised to equal the number:
N= b^{x} (ex. N=10x)
LogbN = X (ex. \log_{10}11=1.04139)
Common logarithm = base of 10
Ex: 100 = 10^2
Log 100 = 2
100 = antilogarithm of 2
Logarithm
Natural logarithms (ln) use the base e, whose value is 2.718282. To relate natural logarithms to common logarithms, the following equation is used:
Laws of Logarithm
See attached image
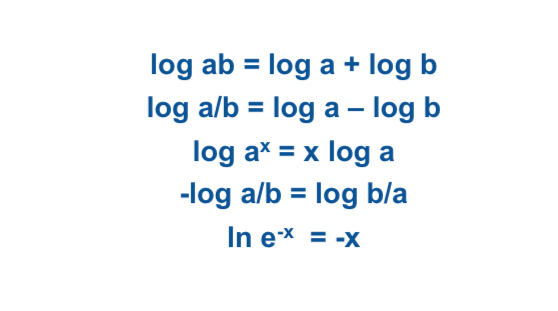
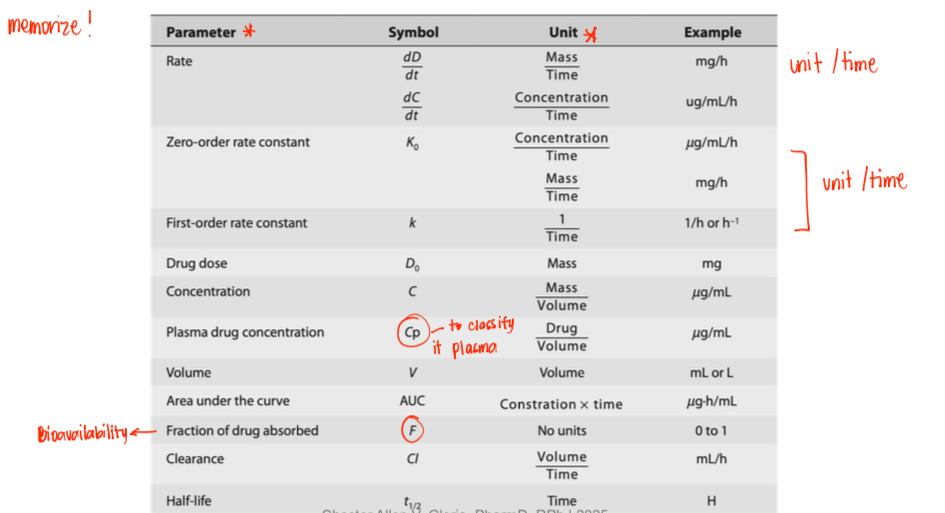
Common Units in Pharmacokinetics
MEMORIZE!!!
Rates and Orders of Reaction
How quickly a drug concentration changes over time
RATE of a chemical reaction
is the velocity with which it occurs.
Change of concentration per unit of time
ORDER of a chemical reaction
is the way in which the concentration of a drug or reactant in a chemical reaction affects the rate.
Zero-order rate process
First-order rate process
How the reactant influences the reaction rate
Rate of Constants
Specific region of the body (plasma or tissue)
Characterize the change of drug concentration in a particular reference region.
Rate of Constants
Give the speed at which a drug:
Enters the compartment (absorption rate constant) ka
Distributes between a central and peripheral compartments (distribution rate constant) kd
Is eliminated from the systemic circulation (elimination rate constant) ke Some drugs, their manner of elimination is either Metabolism or Excretion or both
Rate of Constants
Importance:
If there’s high absorption rate constant = fast onset of action (vice versa)
If you have low distribution rate constant = the drug stay in the lipid rich organ or adipose tissue
If you have low elimination rate constant = slow elimination = the drug will stay in the body longer (toxicity)
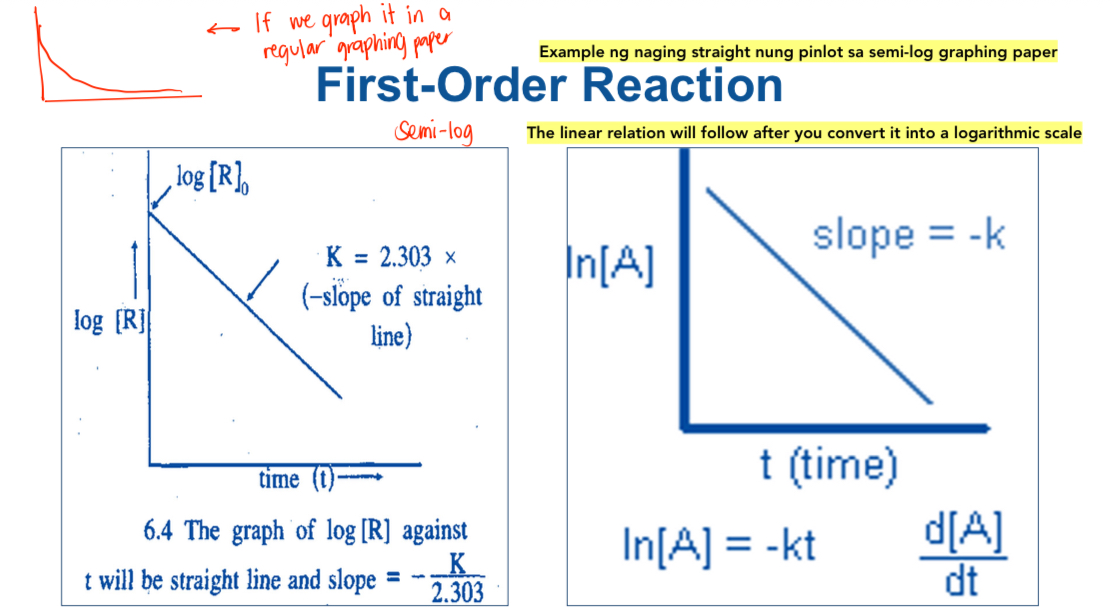
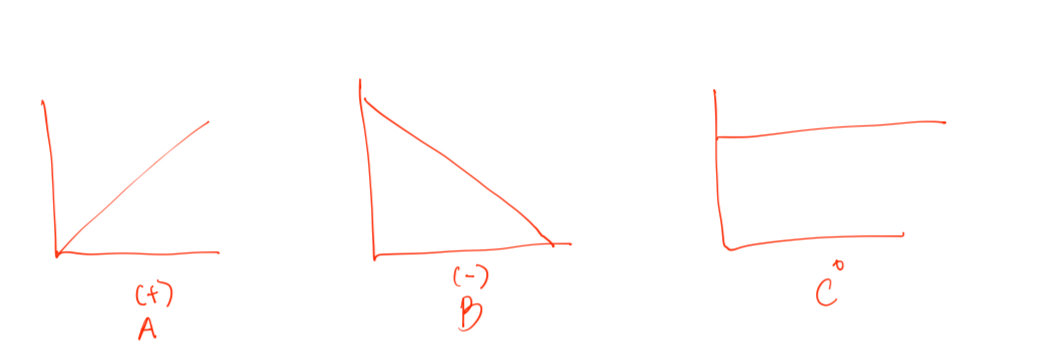
Straight Line
See attached image
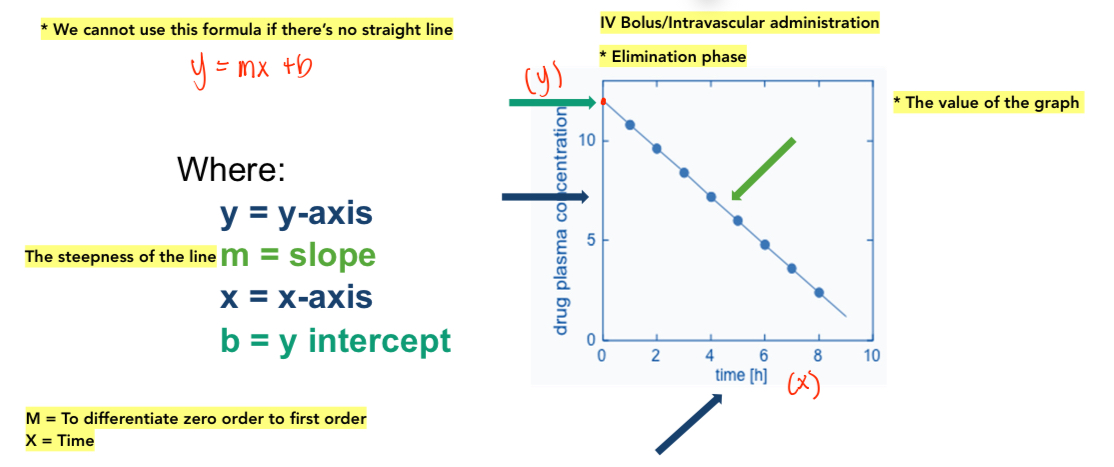
Both Graph A & B has linear relationship
Graph A has positive value (absorption)
Graph B has negative value (elimination)
Graph C no linear relationship
See attached image
Semi-log
(one-sided) plot because only one side/axis is logarithmic.
Zero-Order Reaction
The drug concentration changes with respect to time at a constant rate
C = -k0t + C0
Where:
C = drug concentration at any time
K0 = zero-order rate constant (units of concentration per time) = is the slope of the line
C0 = is the y intercept = drug concentration, when time (t) equals zero
*Negative sign = indicates that the slope is decreasing
Zero-Order Reaction
A straight line results when C is plotted against t
Semilog = non linear
See attached image for normal or regular
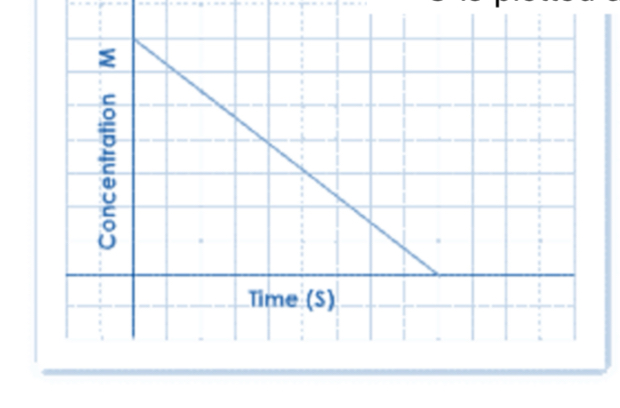
Zero-Order Elimination Kinetics
The plasma concentration vs. time profile during the elimination phase is linear.
Zero-order elimination is rare.
Mostly occurring when the elimination system is saturated.
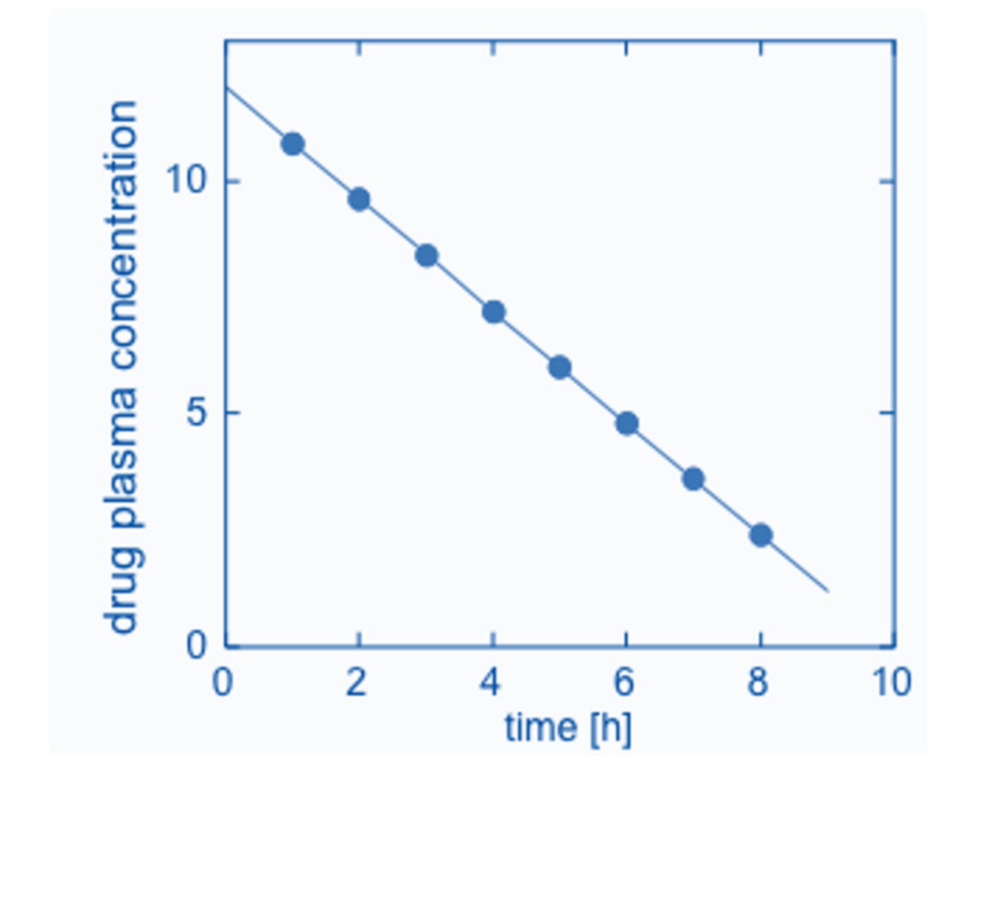
First-Order Reaction
If the zero order is independent to the concentration, the first order is DEPENDENT to conc.
The drug concentration changes with respect to time equal the product of the rate constant and the concentration of drug remaining.
First-Order Reaction
See image below
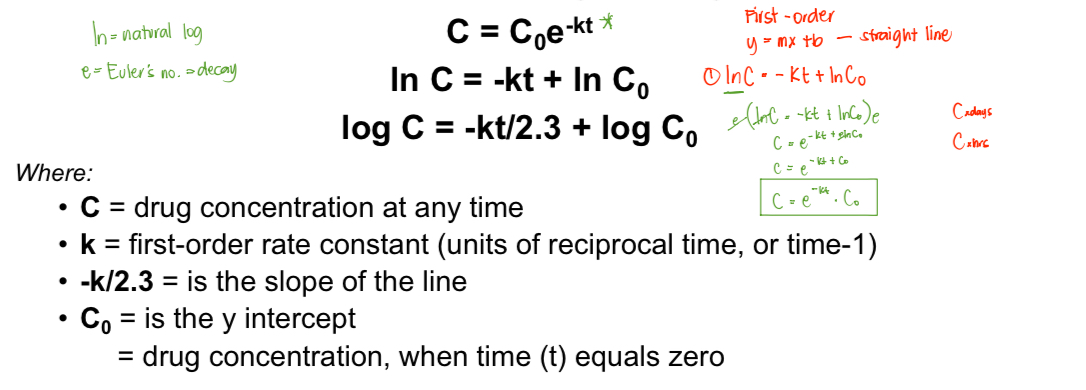
First-Order Reaction
See image below