Lesión celular
1/102
Earn XP
Description and Tags
Capítulo 2
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
103 Terms
Etiología
Causa iniciadora de una enfermedad.
Se agrupa en genéticos (mutaciones hereditarias) o ambientales (infecciosas).
Patogenia
Secuencia de procesos moleculares, bioquímicos y celulares que conducen al desarrollo de una enfermedad.
Explica como las etiologías subyacentes producen las manifestaciones morfológicas y clínicas de la enfermedad.
Cambios morfológicos
Modificaciones estructurales de las células o tejidos característicos de una enfermedad y diagnósticas de un proceso etiológico.
Manifestaciones clínicas
Son producidas por los cambios genéticos, bioquímicos y estructurales en las células y los tejidos.
Son anomalías funcionales que producen signos y síntomas y su progresión.
Adaptaciones
Respuestas funcionales y estructurales reversibles ante cambios en estados fisiológicos y estímulos patológicos, donde se alcanzan estados de equilibrio nuevos que permiten a la célula sobrevivir y seguir funcionando.
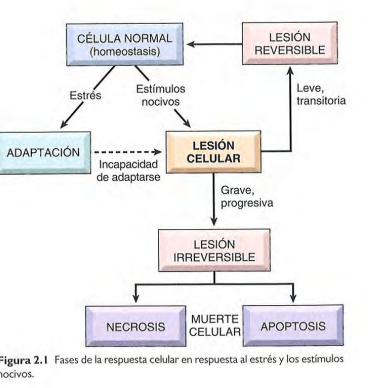
Respuesta adaptativa
Puede ser:
-Hipertrofia: aumento del tamaño
-Hiperplasia: aumento del número de células
-Atrofia: Reducción del tamaño y actividad metabólica de las células.
-Metaplasia: cambio en el fenotipo de las células.
Lesión celular
Secuencia de procesos generada cuando las células agredidas son privadas de nutrientes esenciales o alteradas por mutaciones que afecten a funciones celulares esenciales.
-Puede ser reversible o irreversible.
-En caso de ser irreversible y avanzar puede haber muerte celular.
Muerte celular excesiva
Es resultado de la lesión progresiva.
Puede deberse a diversas a isquemia, infección y tóxicos.
Vías de muerte celular
Necrosis y Apoptosis
¿Qué desencadena la privación de nutrientes?
La autofagia (respuesta celular adaptativa)
Alteraciones metabólicas y lesión crónica.
Se asocian a acumulaciones intracelulares de lípidos, proteínas e hidratos de carbono.
*Puede depositarse calcio en zonas de muerte celular =calcificaciones patológicas.
Hipoxia
Es la deficiencia de oxígeno y causa lesión celular al reducir la respiración oxidativa aeróbica.
-Causas: isquemia, oxigenación inadecuada de la sangre por insuficiencia respiratoria y menor capacidad de transporte de oxígeno en sangre (anemia, intoxicación por CO2, hemorragias graves)
Agentes físicos
Pueden causar lesión celular los traumatismos mecánicos, temperaturas extremas, cambios bruscos de presión atmosférica, etc.
Sustancias químicas
Pueden producir lesión celular. Ej: glucosa o sal en soluciones hipertónicas.
Causan lesión de manera directa o alterando el equilibrio hidroeléctrico en las células.
Agentes infecciosos
Agentes biológicos como rickettsias, bacterias, hongos y formas superiores de parásitos pueden causar daño celular.
Reacciones inmunitarias
Pueden causar lesión celular, como aquellas donde autoantígenos endógenos son responsables de enfermedades autoinmunitarias.
Anomalías genéticas
Por cromosomas extra— síndrome de Down
Sustitución de un par de bases — anemia drepanocítica
Pueden causar lesión celular por una función deficiente de las proteínas.
Desequilibrios nutricionales
Son causas de lesión celular.
Ej. deficiencias de proteínas y calorías, escases de vitaminas, anorexia, bulimia u obesidad.
Cambios morfológicos de lesión celular
Son observables con métodos de estudio sensibles (técnicas histoquímicas, ultraestructurales o bioquímicas)
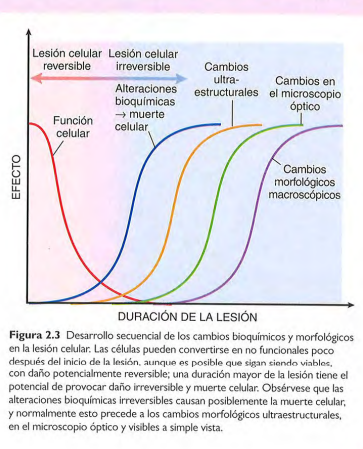
Lesiones secundarias en lesión celular
Aparecen de horas a días después, son observables en el microscopio óptico
Isquemia del miocardio
La tumefacción celular es un cambio morfológico reversible producido en minutos y que indica daño celular mantenido.
Si progresa de 1 a 2 hrs puede ser irreversible.
Visible en M.O indicios de muerte celular de 4-12hrs después.
Características de la necrosis y la apoptosis
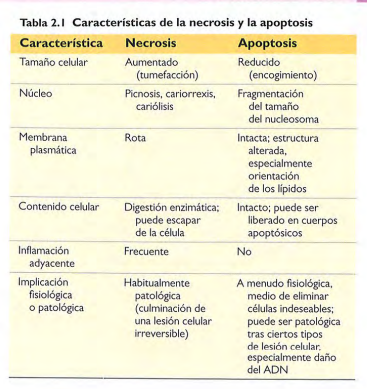
Lesión celular reversible
Presenta alteraciones funcionales y estructurales en estadios iniciales o formas leves de lesión, corregibles si se elimina el estímulo dañino.
Primeras alteraciones en lesión reversible
-Tumefacción generalizada de la célula y sus orgánulos.
-Formación de vesículas en membrana plasmática
-Separación de ribosomas en RE
-Agregación de cromatina nuclear
¿A qué se debe la tumefacción celular?
Por la entrada de agua a las células, por el fracaso de la bomba de Na+-K+ dependiente de ATP de la membrana plasmática en relación con una deficiencia de oxígeno que interfiere en la fosforilación oxidativa mitocondrial o una lesión mitocondrial secundaria a radiación o tóxicos.
Cambio graso
En órganos activos en el metabolismo de lípidos, es causado por un daño tóxico que altera las vías metabólicas y provoca acumulación rápida de vacuolas lipídicas con triglicéridos.
¿Cuál es la primer manifestación de lesión celular?
Tumefacción celular.
Causa palidez, mayor turgencia y aumento de peso del órgano afectado.
Cambio hidrópico/ degeneración vacuolar
Patrón de lesión no mortal donde se aprecian vacuolas transparentes en el citoplasma; representan segmentos distendidos y desprendidos del RE.
Necrosis
Proceso patológico consecuencia de una lesión celular grave.
Producida por isquemia, exposición a toxinas, quemaduras, daños físicos y químicos, *por proteasas activas (pancreatitis).
Características de la necrosis
Desnaturalización de proteínas celulares, escape del contenido celular por las membranas dañadas, inflamación local y digestión enzimática de la célula herida mortalmente.
¿Qué produce la inflamación en la necrosis?
El contenido celular que escapa a través de la membrana plasmática dañada al espacio extracelular, donde incita la inflamación.
Patrones moleculares asociados a daño (DAMP)
Sustancias liberadas por las células lesionadas.
-ATP (de las mitocondrias)
-Ácido úrico (de la degradación del ADN)
Estos son reconocidos por receptores en macrófagos, desencadenando fagocitosis de los restos, producción de citocinas (inflaman)
La fagocitosis y la digestión enzimática eliminan las células necróticas.
¿Qué permite el escape de proteínas intracelulares en la necrosis?
La identificación célular específica de tejido mediante análisis de sangre.
Corazón —troponina
Epitelio del conducto biliar — fosfatasa alcalina
Hepatocitos — transaminasas
¿Qué tiempo toma la detección de troponinas cardíacas séricas?
2 horas después de la necrosis de células miocárdicas.
*Para tratamiento de infarto de miocardio.
Fenómenos que caracterizan la lesión irreversible
-Incapacidad de revertir la disfunción mitocondrial (ausencia de fosforilación oxidativa y generación de ATP)
-Alteraciones graves de la función de membrana.
Cambios nucleares en apoptosis
Se deben a la degradación del ADN.
-Cariólisis: reducción de la basofilia de la cromatina, refleja pérdida de ADN secundaria a degradación enzimática por endonucleasas.
-Picnosis: reducción el tamaño del núcleo y mayor basofilia. La cromatina se condensa en una masa basófila contraída.
-Cariorrexis: el núcleo picnótico sufre fragmentación. 1-2 días después desaparece por completo.
Necrosis coagulativa
La arquitectura del tejido muerto se mantiene, el tejido tiene una textura firme.
- Desnaturaliza proteínas estructurales y enzimas bloqueando la proteólisis de células muertas.
-Las células necróticas son degradadas por enzimas lisosómicas liberadas por leucocitos infiltrantes, por fagocitosis.
-Puede ser causada por obstrucción de un vaso (menos en encéfalo)
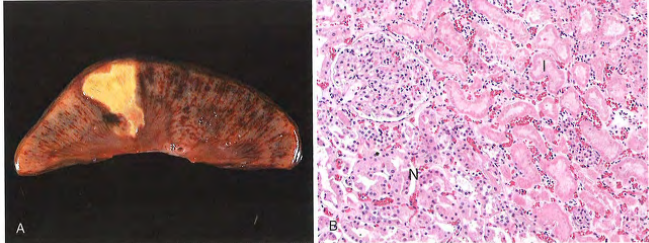
¿Cómo se denomina un área localizada de necrosis coagulativa?
Infarto
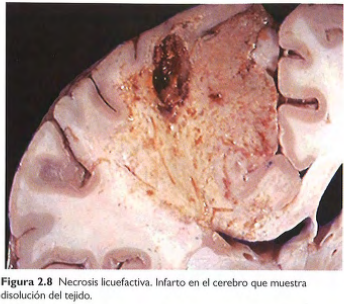
Necrosis licuefactiva
-Caracterizada por digestión de células muertas, transforma el tejido en un líquido viscoso.
-En infecciones bacterianas focales y fúngicas (Los microbios estimulan los leucocitos y su liberación de enzimas)
-Hay pus (material amarillo con leucocitos)
-En muerte celular de SNC.
Necrosis gangrenosa
-Se aplica a extremidades que se quedan sin vascularización y han sufrido necrosis (coagulativa) en múltiples planos tisulares.
-Al superponerse una infección bacteriana existe necrosis licuefactiva dando lugar a gangrena húmeda.
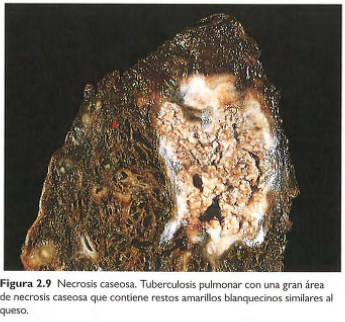
Necrosis caseosa
-En focos de infección tuberculosa
-Tiene aspecto friable y color blanco. (Parecido a queso)
-Se almacenan células fragmentadas o lisadas y restos granulares amorfos delimitados por un margen inflamatorio definido (granuloma)
Necrosis grasa
-Áreas focales de destrucción de grasa, por liberación de lipasas pancreáticas activadas al interior del páncreas y la cavidad peritoneal. (En pancreatitis aguda)
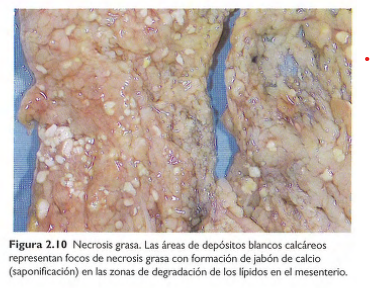
¿Qué ocurre en la pancreatitis aguda?
-Las enzimas pancreáticas salen de las células acinares dañadas y licuan las membranas de las células adiposas del peritoneo, liberando ésteres de triglicéridos que son escindidos por lipasas pancreáticas.
-Se generan ácidos grasos que se combinan con calcio (saponificación de la grasa)
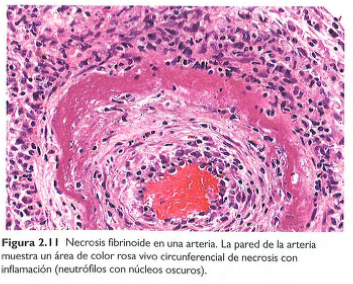
Necrosis fibrinoide
-Se observa en reacciones autoinmunitarias que afectan los vasos sanguíneos.
-Se da al depositarse imunocomplejos con las proteínas citoplasmáticas que salen de los vasos.
-Se observan fibrinoides (rosa brillante y amorfa en H-E)
Lesión celular reversible
Características:
Tumefacción celular, Cambio graso ,Vesículas ,Pérdida de microvellosidades de la membrana plasmática ,Tumefacción de las mitocondrias ,Dilatación del RE, Eosinofilia
Necrosis
Proceso patológico donde se destruyen las membranas celulares, salen enzimas y se induce inflamación local para retirar las células dañadas.
Características morfológicas:
Eosinofilia, disminución de tamaño, fragmentación y disolución del núcleo, descomposición de la membrana plasmática y membranas de los orgánulos, figuras de mielina, escape y digestión enzimática del contenido celular.
Apoptosis
Muerte celular inducida por un programa de suicidio regulado, donde las células destinadas a morir activan enzimas intrínsecas que degradan su ADN genómico y las proteínas nucleares y citoplásmicas.
Cuerpos apoptósicos
Son células apoptóticas descompuestas en fragmentos rodeados por membrana plasmática.
Contienen parte del citoplasma y núcleo.
Causas de la apoptosis
-Procesos fisiológicos normales
-Mecanismo fisiopatológico de pérdida celular.
Retirada de células supernumerarias
La muerte celular es esencial para involución de estructuras primordiales y remodelación de tejidos en maduración.
Involución de tejidos dependientes de hormonas al desparecer estas
La apoptosis ayuda con la degeneración de células endometriales en ciclo menstrual, atresia de folículos ováricos en menopausia y regresión de mama tras destete.
Recambio celular en poblaciones de células en proliferación
La apoptosis ayuda con:
-Linfocitos inmaduros de médula ósea y rimo
-Linfocitos B de centros germinales que no expresen receptores de antígenos útiles
-Células de criptas intestinales
*Ayuda a mantener la homeostasis de las células.
Eliminación de linfocitos autorreactivos potencialmente peligrosos
La apoptosis ayuda a prevenir reacciones inmunitarias contra tejidos propios
Muerte de células que completaron su objetivo útil
La apoptosis de neutrófilos luego de una respuesta inflamatoria aguda y linfocitos al final de respuesta inmunitaria.
Apoptosis por daño del ADN
Radiación y fármacos citotóxicos antineoplásicos dañan al ADN, por radicales libres.
En caso de no corregirse por mecanismos de reparación se induce la apoptosis, cuyo efecto es proteger al impedir la supervivencia de las células con mutaciones de ADN.
Apoptosis por acumulación de proteínas mal plegadas
Muerte celular por proteínas intracelulares plegadas mal y respuesta al estrés del RE.
Apoptosis durante infecciones
Sobre todo en víricas, como resultado del propio virus o la respuesta inmunitaria del huésped.
La respuesta del huésped ante los virus depende de los CTL específicos frente a proteínas víricas (genera apoptosis de células infectadas) intentando eliminar reservorios de infección.
Apoptosis por atrofia patológica de órganos parenquimatosos tras obstrucción de conductos
En páncreas, glándula parótida y riñón.
Características de la apoptosis
Reducción del tamaño celular, condensación de la cromatina, formación de vesículas citoplásmicas y cuerpos apoptósicos y fagocitosis de las células apoptósicas o cuerpos celulares por macrófagos.
Reducción del tamaño celular
El tamaño de la célula disminuye, el citoplasma es denso y eosinófilo.
-Orgánulos más agregados
Condensación de la cromatina
DISTINTIVO DE LA APOPTOSIS
La cromatina se agrega periféricamente bajo la membrana nuclear, en masas.
El núcleo se rompe en dos o más fragmentos.
Formación de vesículas citoplásmicas y cuerpos apoptósicos
La célula apoptósica muestra vesículas por toda la membrana, después se fragmentan las células muertas en cuerpos apoptósicos rodeados por membrana, compuestos por citoplasma y orgánulos densamente empaquetados con o sin fragmentos de núcleos.
Fagocitosis de células apoptósicas o cuerpos celulares por macrófagos
Los cuerpos apoptósicos son ingeridos por fagocitos y degradados por enzimas lisosómicas del fagocito.
¿Quién produce la apoptosis?
La activación de las enzimas caspasas (presentes en forma de proenzimas inactivas que para activarse necesitan una ecisión enzimática.
Fases de la apoptosis
Fase de iniciación: las caspasas se activan catalíticamente y desatan una cascada de otras caspasas.
Fase de ejecución: las caspasas terminales activan la fragmentación celular.
Vías que convergen en la actividad de caspasas
-Vía mitocondrial
-Vía del receptor de muerte
Vía mitocondrial (Intrínseca de la apoptosis)
Es la responsable de la apoptosis.
Es resultado de la mayor permeabilidad de la membrana externa mitocondrial, con liberación de moléculas proapoptósicas del espacio intermembrana de la mitocondria al citoplasma.
La mitocondria contiene citocromo C que al liberarse al citoplasma inicia la apoptosis.
¿Quién determina la liberación de proteínas proapoptósicas?
La membrana externa de la mitocondria, controlada por la familia de proteínas BCL2 (clasificadas en 3 grupos según su función proapoptósica o antiapoptósica y los dominios de homología BCL2 “BH” que poseen)
BCL2 antiapoptósicas
BCL2, BCL-XL y MCLl.
-Tienen 4 dominios BH (BH1-4)
-Se sitúan en la membrana externa de la mitocondria, citosol y membranas del RE.
-Mantienen impermeable la membrana externa mitocondrial, impidiendo fuga de citocromo C y proteínas inductoras de muerte al citosol.
BCL2 Proapoptósicas
BAX y BAK.
-Contienen los primeros 3 dominios BH (BH1-3)
-Al activarse BAX y/o BAK se oligomerizan dentro de la membrana externa de la mitocondria y aumentan su permeabilidad.
-BAX y BAK forman un canal en la membrana mitocondrial externa que permite fuga de citocromo c del espacio intermembrana.
BLC2 iniciadores de la apoptosis regulada
BAD, BIM, BID, Puma y Noxa
-Tienen un único dominio BH, el tercero de los cuatro dominios BH (por ello reciben solo BH3)
-Su actividad esta modulada por sensores de estrés y daño celular.
-Cuando se regulan al alza y activan pueden iniciar apoptosis.
¿Cómo se protegen las células de la apoptosis?
Factores de crecimiento y señales de supervivencia estimulan la producción de proteínas antiapotósicas (BCL2)
Proteínas solo BH3
Se regulan al alza por aumento de su transcripción y/o modificaciones tras la traducción (Ej.Fosforilación)
-Activan BAX y BAK que forman oligómeros que se insertan en la membrana interna de la mitocondria, permiten que las proteínas de la membrana salgan al citoplasma.
-BH3 se une a BCL2 y y BCL-XL bloqueando la función de BAX y BAK.
¿Cuál es el resultado de activar BAX-BAK y la pérdida de funciones de BCL2?
La liberación en el citoplasma de proteínas mitocondriales (citocromo C) capaces de activar la cascada de caspasas.
¿Qué pasa con el citocromo C liberado en el citosol?
Se une a APAF-1 y se forma un apoptosoma que se une a la caspasa 9 (iniciadora esencial de la vía mitocondrial) y promueve su escisión autocatalítica generando formas catalíticamente activas de la enzima.
Caspasa 9 activa desencadena una cascada de activación de caspasas escindiendo y activando otras caspasas (caspasa 3) que medían la fase de ejecución de la apoptosis.
Smac/DIABLO
Proteína mitocondrial que pasa al citoplasma, uniéndose a proteínas citoplásmicas que funcionan como inhibidores fisiológicos de la apoptosis (IAP) y las neutralizan.
Función de los IAP
Bloquean la activación inapropiada de las caspasas como caspasa 3 y mantiene vivas las células.
Su inhibición permite iniciar la cascada de caspasas.
Vía extrínseca de la apoptosis (iniciada por el receptor de la muerte de la apoptosis)
Inicia con la ocupación de los receptores de muerte de la membrana plasmática.
Receptores de muerte
Miembros de la familia del factor de necrosis tumoral (TNF), contienen un dominio citoplásmico implicado en interacciones entre proteínas.
Su dominio de muerte hace llegar señales apoptósicas.
*Algunos TNF no tienen dominio de muerte, sino que activan cascadas inflamatorias.
-TNFR1 y CD95 inducen la apoptosis por Fas.
-El Fas L se une a fas y se encuentra en linfocitos T que reconocen autoantígenos y en algunos CTL destruyen células infectadas por virus y tumorales.
¿Qué sucede cuándo FasL se une a Fas?
Se agrupan tres o más moléculas de Fas y sus dominios de muerte citoplásmicos forman un lugar de unión para FADD.
Ya fijo al complejo, FADD se une a caspasa 8 inactiva agrupando múltiples caspasa y provocando escisión autocatalítica y generación de caspasa 8 activa.
Función de caspasa 8 activa
Inicia la misma secuencia de caspasas ejecutoras que la vía mitocondrial.
¿Quién inhibe la apoptosis mediada por Fas?
FLIP, que se une a procaspasa 8, bloqueando la unión del FADD, pero no es capaz de activar caspasa.
¿Qué caspasa activa la vía mitocondrial intrínseca para la fase final de la apoptosis?
Caspasa 9
¿Qué caspasa activa la vía mitocondrial extrínseca para la fase final de la apoptosis?
Las caspasas 8 y 10.
Función de caspasas 8 y 10 activos
Desencadenan la activación rápida y secuencial de las caspasas ejecutoras (3 y 6) que actúan sobre componentes celulares.
¿Para qué se forman los cuerpos apoptósicos?
Para descomponer las células en fragmentos “come de un bocado” accesibles a los fagocitos.
Fosfatildiserina
-En células sanas está en su hoja interna de la membrana plasmática.
-En células apoptósicas se voltea y expresa en la capa externa de la membrana, donde se reconoce por receptores de macrófagos.
Eferocitosis
Proceso de fagocitosis de células apoptósicas donde C1q recubre los cuerpos apoptósicos para ser reconocidos por fagocitos que se unen a estos y los rodean.
Apoptosis (resumen)
Mecanismo regulado de muerte celular que sirve para eliminar células indeseadas y dañadas irreparablemente, con una reacción mínima posible del huésped.
Características de la apoptosis
-Degradación enzimática de proteínas y ADN.
-Iniciada por caspasas, reconocimiento y eliminación de células muertas por fagocitos.
Vías principales de apoptosis (resumen)
Mitocondrial intrínseca:
-Desencadenada por pérdida de señales de supervivencia, daño del ADN y acumulación de proteínas mal plegadas (estrés del RE)
-Hay fuga de proteínas proapoptósicas de la membrana mitocondrial al citoplasma y activación de caspasas.
-Inhibida por miembros antiapoptósicos de BCL2 (inducidos por señales de supervivencia)
Vía del receptor de muerte (extrínseca) :
-Elimina linfocitos autorreactivos.
-Mecanismo de destrucción de células por linfocitos T citotóxicos, inicia por unión a receptores de muerte.
-Los ligandos responsables pueden ser solubles o expresarse en la superficie de células adyacentes.
Necroptosis
-Muerte celular híbrida que comparte aspectos de necrosis y apoptosis.
-Se caracteriza por pérdida de ATP, tumefacción de la célula y orgánulos, generación de ERO, liberación de enzimas lisosómicas y rotura de la membrana plasmática.
-Desencadenada por unión a TFNR1 y proteínas en ARN y ADN del virus.
-Independiente de caspasas.
-Depende de RIPK1 y RIPK3.
-La señalización por RIPK1-3 condiciona la fosforilación de MLKL que forma poros en la MP.
-Al liberarse los componentes celulares hay reacción inflamatoria.
¿Qué produce la unión de TNFR1 a RIPK1 y RIPK3?
TNFR1 recluta las cinasas como complejo multiproteínico.
RIPK3 fosforila una proteína MLKL la cual en respuesta, hace que sus monómeros se reúnan en oligómeros, pasando del citosol a la membrana plasmática y provocando la rotura de la membrana plasmática.
¿En qué estados patológicos se asocia con muerte celular la Necroapoptosis?
Esteatohepatitis, pancreatitis aguda, lesión por isquemia-reperfusión, enfermedades neurodegenerativas (Parkinson)
Piropiptosis
Forma de apoptosis acompañada de liberación de IL-1 (inductora de fiebre)
-Los productos microbianos que entran son reconocidos por receptores citoplásmicos inmunitarios innatos y activan el inflamasoma.
-Deoende de activacióin de caspasa 1, que escinde el precursor de IL-1 para formar IL1 activa.
-Este mecanismo mata células infectadas y desencadena inflamación local.
Inflamasoma
Complejo multiproteínico que activa la caspasa 1 (enzima conversora de IL-1B que escinde el precursor de IL-1 y libera su forma activa.
Ferroptosis
-Muerte celular que se activa cuando cantidades extracelulares excesivas de hierro o EROS superan las defensas antioxidantes dependientes del glutatión causando peroxidación de los lípidos de la membrana.
¿Qué puede producir la peroxidación de los lípidos?
Alteración en funciones de la membrana:
-Fluidez de la membrana
- Interacciones entre lípidos y proteínas.
-Transporte de iones y nutrientes
-Vías de señales
EFECTO GLOBAL: pérdida de la permeabilidad de la membrana plasmática.
-Relacionada con cáncer, enfermedades neurodegenerativass y accidente cerebrovascular.
Autofagia
-La célula consume su propio contenido.
-Hay transporte de materiales citoplásmicos a lisosomas para su degradación.
-Mecanismo de supervivencia cuando hay privación de nutrientes.
-Implicada en envejecimiento y ejercicio y procesos patológicos.
PASOS:
1) NUCLEACIÓN Y FORMACIÓN DE MEMBRANA DE AISLAMIENTO (FAGÓFORO)
2) FORMACIÓN DEL AUTOFAGOSOMA DE LA MEMBRANA DE AISLAMIENTO (Dentro queda orgánulos intracelulares y estructuras del citosol)
3) Maduración del autofagosoma por fusión con los lisosomas, para suministrar enzimas digestivas que degradan el contenido del autofagosoma.
ATGS
-Genes relacionados con la autofagia.
-Sus productos son necesarios para creación del autofagosoma.
-La privación de nutrientes o agotamiento de factores de crecimiento activan un complejo que promueve reclutamiento de ATGS para formar núcleo de membrana de iniciación.
-La membrana de iniciación rodea y captura su carga citosólica, cerrándose para formar el autofagosoma.
¿A qué se debe el alargamiento y cierre de la membrana de iniciación?
r= a dos sistemas de conjugación que condicionan el enlace covalente del lípido fosfatidiletanolamina (PE) con la cadena ligera de la proteína asociada a microtúbulos 3 (LC3)
-El autofagosoma se fusiona con lisosomas para formar un autofagolisosoma.
*Al final la membrana interna y la carga citosólica rodeada son degradadas por enzimas lisosómicas.