SNP, GWAS, and post-GWAS
1/38
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
39 Terms
Genome wide association studies (GWAS)
Identify associations between genetic variations (loci) and traits (including diseases).
Test for differences in the frequency of genetic variants between individuals who are ancestrally similar but differ phenotypically.
Genetic variations:
Single nucleotide polymorphism (SNP) → most common.
Copy number variants.
Large sequence variations.
Sinlge nucleotide polymorphism (SNP)
Most common genetic variation (~90% of human variation).
Single base pair change (substitution, insertion, or deletion).
Location:
Coding: may change protein sequence.
Non-coding (majority): may affect gene expression, timing, or location.
SNP database
NCBI Short Genetic Variation database (dbSNP) catalogs short variations in nucleotide sequences for humans.
Major and minor allele
Major allele is the most common variant found in a population, while the minor allele is the less common or rarer variant at that same position.
Minor allele frequency (MAF).
MAF > 1% → common SNP.
MAF < 1% → rare SNP.
Major and minor alleles are population-specific.
Focus often on the minor allele because minor alleles are crucial for identifying disease risks and studying genetic selection.
Synonymous vs. nonsynonymous SNPs
Synonymous SNPs:
Do not change the amino acid sequence of a protein.
Silent change.
Nonsynonymous SNPs:
Potentially alter protein structure and function.
This distinction arises because the genetic code is redundant, with multiple codons sometimes coding for the same amino acid.
Missense vs. nonsense SNPs
Missense and nonsense SNPs are both nonsynonymous mutations.
Change the amino acid sequence.
Missense mutation:
Results in a different amino acid being incorporated into the protein.
It will alter the protein’s structure and function.
Nonsense mutation:
This changes from a sense codon to a premature stop codon.
This leads to a truncated and often non-functional protein.
Linkage disequilibirum (LD) blocks
LD = non-random association of genes.
There is a pattern.
Sets of nearby SNPs on the same chromosome are inherited together in blocks.
Haplotype/LD block:
A group of alleles that are co-inherited as a single block.
LDTools: LDHap, LDMatrix, etc.
Tag SNPs
A few SNPs are enough to identify the haplotypes in a block uniquely.
Haplotype: a group of alleles/variants on the same chromosome that are inherited together.
Reduce the number of SNPs required to examine the entire genome for association with a phenotype.
Methods other than measure SNPs
Genotyping: targets known SNPs at specific sites (e.g., SNP arrays), cheaper, limited to pre-selected variants.
DNA sequencing: reads entire DNA, finds known & novel SNPs (via variant calling → VCF), more data, more expensive.
How to do SNP selection (genotyping)
Identify DNA regions of interest.
Identify patterns of SNPs that are inherited together on a chromosome.
HapMap project: tested the association of millions of SNPs across multiple populations → produced the SNP panels that are used today.
Select tag SNPs that can represent a block of associated SNPs.
SNP genotype models
SNP genotypes can be coded differently based on genetic models.
Additive model (ADD) → commonly used.
Dominant model (DOM).
Recessive model (REC).
SNP genotypes are commonly coded as 0, 1, or 2 to represent the number of copies of the minor allele.
0 → homozygous dominant (e.g., AA).
1 → heterozygous (e.g., Aa).
2 → homozygous recessive (e.g., aa).

Genotype/haplotype phasing
Current technology gives genotypes but not haplotypes.
The process of determining which alleles are located on the same chromosome, i.e., the haplotype.
Phasing tools: SHAPEIT5, BEAGLE5.5.
GWAS association tests
Chi-square / case-control: simple test, no confounders.
No confounders = assumes no other factors (e.g., age, ancestry) affect the association between a variant and a trait.
Linear regression: continuous traits (height, BMI, blood pressure).
Logistic regression: binary traits (disease yes/no).
Linear mixed models: account for relatedness among individuals.
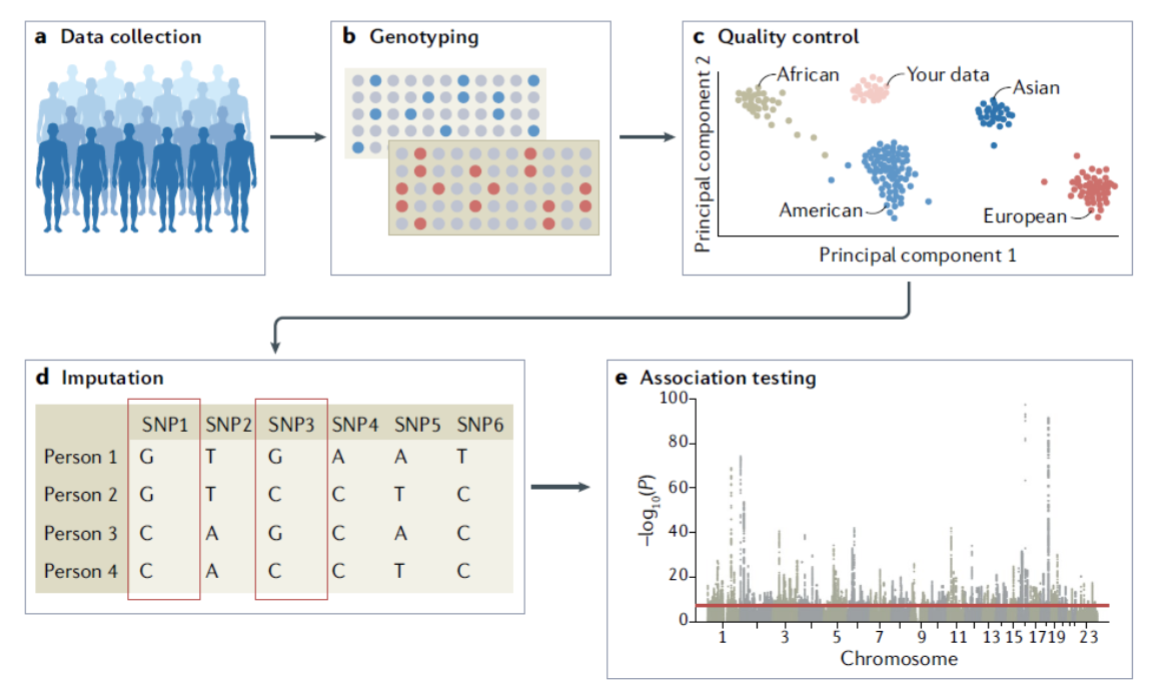
GWAS example
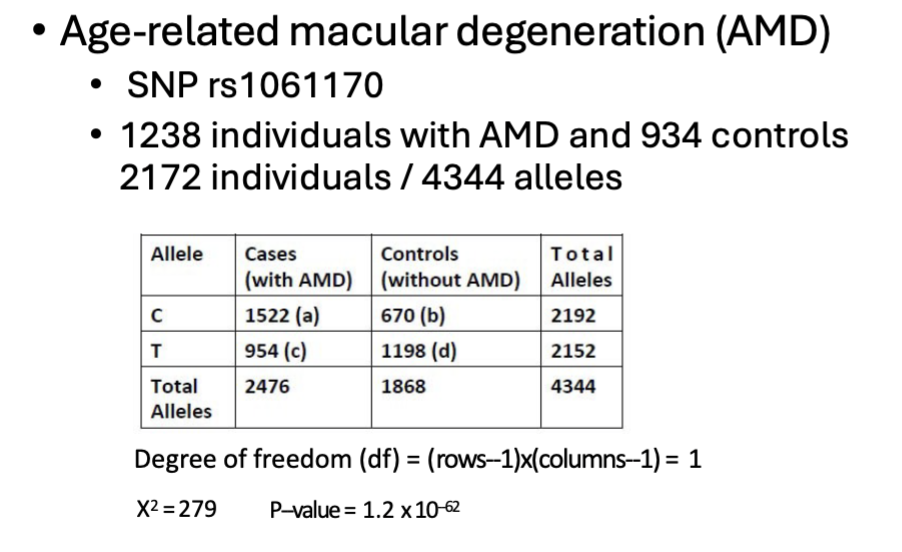
Odds ratio
Measure of effect size.
OR = 1, no disease association.
OR > 1, allele C increases risk of disease.
OR < 1, allele C decreases the risk of disease.

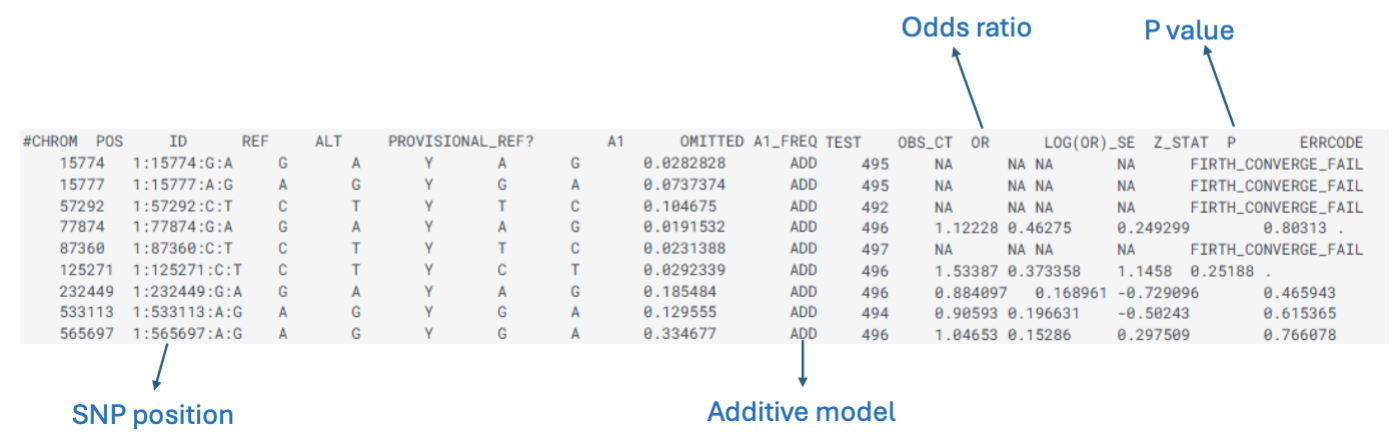
PLINK
A software tool for analyzing genetic data, especially for GWAS, including association testing, quality control, and data management.
Mostly shared.
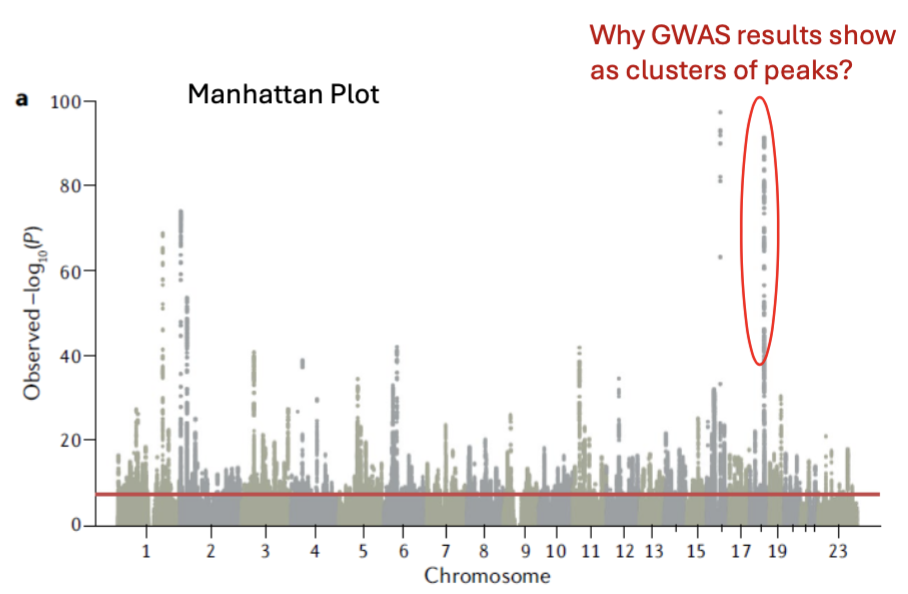
How do we visualize GWAS results
Manhattan plot:
Bonferroni testing threshold of p < 5 × 10-8.
Multiple test correction.
Genome-wide significance threshold = red line on the image.
The peaks are SNPs that are close to each other = co-inherited.
Significance threshold varies depending on:
Number of SNPs examined.
Number of subjects included.
Minor allele frequencies.
Etc.
LocusZoom: a combination of a Manhattan plot and a genome browser.
Genotype datasets for large scale GWAS
Biobanks and large population-based studies with genetic and phenotype data available for research.
For USA → ‘All of Us’ initiative or 23andMe.
Genotype data are typically restricted due to re-identification risk.
Application needed.
Databases for GWAS summary statistics
GWAS Catalog.
GWAS Atlas.
Allow easy access to summary statistics for thousands of traits.
Many downstream analyses are built on the summary statistics, rather than the raw genotype itself.
Post-GWAS analysis
Functional mapping:
Where are they located in the DNA?
Pathways enriched by GWAS findings.
How may they influence molecular functions leading to disease?
Identify the tissues or cell types where these variants are likely to act.
Causal variants identification:
GWAS findings come up as clusters → highly correlated.
Which one is likely causal?
Polygenic risk score.
Functional mapping of GWAS findings - Where are they located in the DNA?
Chromosome & base pair position.
Coding regions: synonymous / nonsynonymous.
Non-coding regions (majority): introns, promoters, enhancers, histone marks, DHS sites.
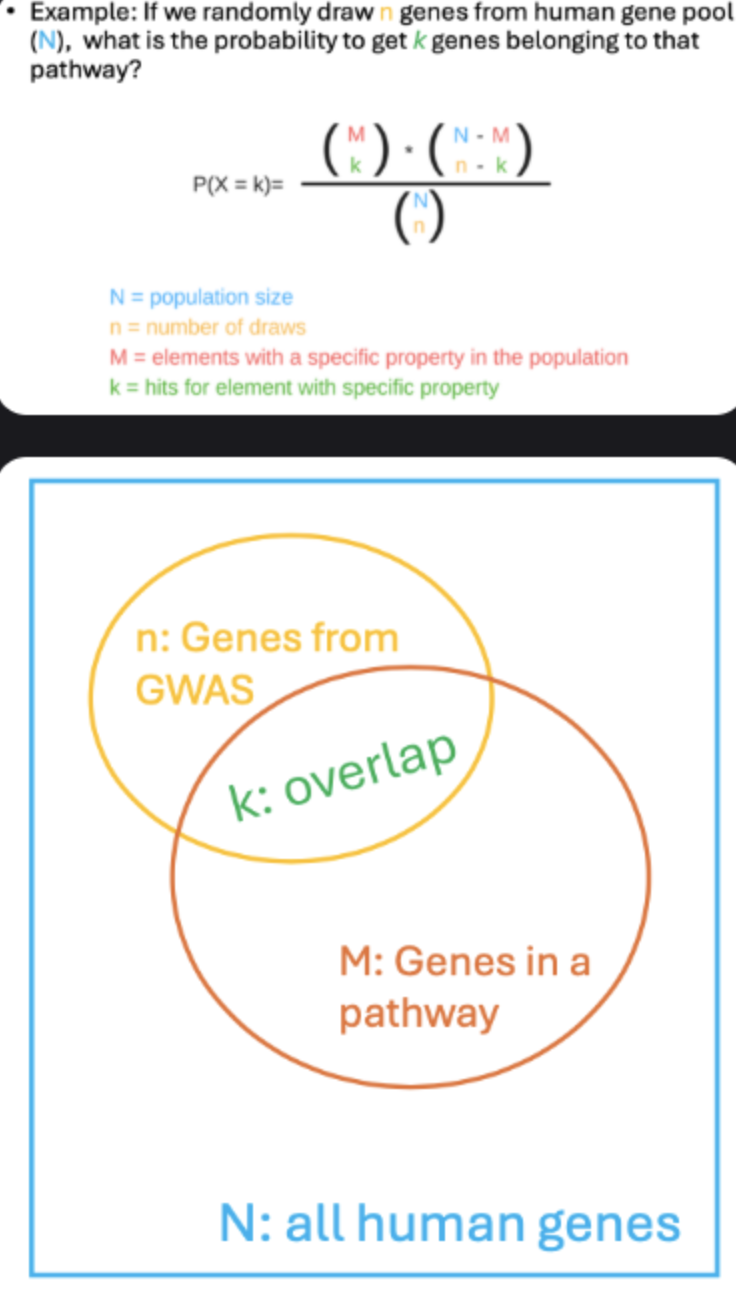
Pathway analysis: link nearest genes to biological pathways; test overlap significance with hypergeometric test (e.g., via EnrichR).
Functional mapping of GWAS findings - How may they influence molecular functions leading to disease?
eQTL: SNP affecting gene expression.
Other QTLs: pQTL, methylation QTL, etc.
Link DNA → gene expression → molecular/phenotypic traits → disease risk.
Functional mapping of GWAS findings - Which tissues or cell types these variants are likely ot act on?
GWAS SNPs: eQTLs are tissue- and/or cell-specific.
Nearest genes of GWAS SNPs.
GTEx project
Studies how genetic variation affects gene expression in normal human tissues.
Focuses on nearby (cis) eQTLs.
eGene: gene whose expression is significantly influenced by ≥1 nearby eQTL.
Functional mapping of GWAS findings - Causal variants identification
Fine-mapping is a statistical analysis that identifies the causal variant(s) within a GWAS locus for a disease.
CausalDB.
Fine mapping steps
Define locus: pick region of interest from GWAS.
Expand region: include variants in LD with lead SNP.
Gather data: association stats (z-scores/SE), LD info.
Identify causal variants: heuristic LD, penalized regression, Bayesian models.
Heuristic LD approach
Pick the lead SNP + group nearby correlated SNPs based on LD thresholds.
High LD = inherited together.
Penalized linear regression approach
Penalize the regression to avoid overfitting when SNPs are numerous and correlated.
Only a few SNPs within a region are causal.
Bayesian models
Posterior inclusive probability (PIP): probability a SNP is causal given data/model.
Credible set: variants whose cumulative PIP reaches confidence threshold (e.g., 95%).
Goal: make credible set as small as possible.
Tools: CAVIAR, FINEMAP, SuSiE, PAINTOR.
Heritability
Measures how much of the variation in a trait is due to genetic differences.
A heritability of 0.7 means that genetic factors explain 70% of the variation in that trait.
Can either be:
Twin-based:
Gold standard for estimating the broad-sense heritability.
GWAS-based.
SNP-based.
Twin-based heritability
Compare similarity of traits in relatives.
Closer relatives share more genes: MZ (monozygotic) twins ~100%, DZ (dizygotic) twins ~50%.
If MZ similarity ≫ DZ similarity → trait largely genetic.
ACE model for twin based heritability

GWAS-based heritability
Definition: proportion of trait variance explained by genotyped SNPs.
From genotype data: use GRM (Genetic Relationship Matrix) → tool: GCTA.
GRM is based on pairwise genetic similarities between individuals.
From GWAS summary stats: regress SNP test stats (χ²) on LD scores → tool: LDSC.
Missing heritability
Definition: portion of trait variance not captured by GWAS.
Due to: rare variants (mendelian variants), structural variants, small-effect common SNPs (polygenic risk scores).
Genome-wide Complex Trait Analysis (GCTA)
Estimates heritability explained by all common SNPs simultaneously.
Finds that common SNPs collectively explain a substantial portion of trait heritability.
Explains more than GWAS hits but less than twin-based estimates.
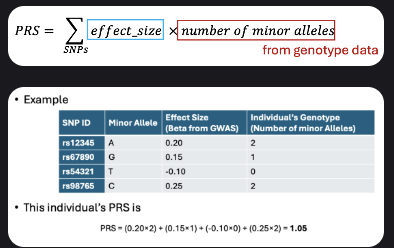
Polygenic risk score
Definition: summarizes overall genetic risk for a trait/disease in a single value.
Combines effects of many variants; each contributes a small amount.
Metrics: association (p-value), variance explained (R²), effect size (β/OR), discrimination (AUC).
Tools: LDPred, PRSice.
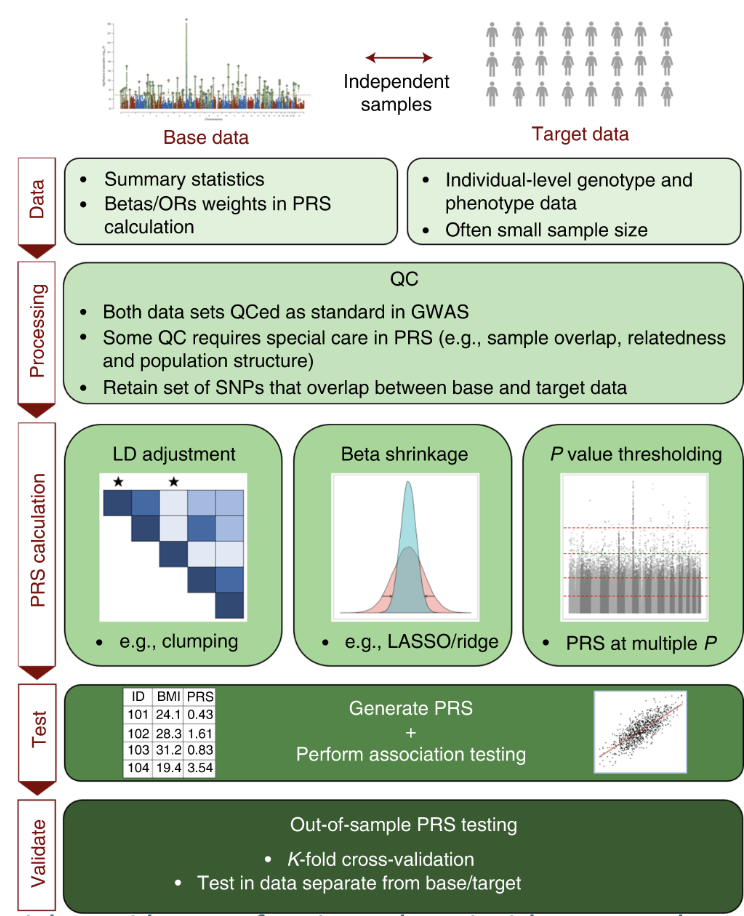
Fundamental steps of a PRS analysis
Base & target QC: check GWAS heritability, different populations, allele alignment, sample size, genome build.
PRS calculation: control for LD, adjust for inflated GWAS effect sizes.
PRS testing & validation.
Interpreting PSR
Can convert PRS to z-scores or percentiles for comparison.
Top decile → higher disease risk vs. bottom decile.
Probabilistic: high PRS ≠ guaranteed disease, low PRS ≠ protection.
PSR applications
Applications: patient stratification (grouping patients based on risk), prevention, trials.
Limits: partial heritability, ancestry bias, ignores interactions (gene-gene interactions), GWAS-dependent (power depends on GWAS sample size and quality).