Human Genetics 1, 2, 3 + 4
1/64
Earn XP
Description and Tags
(done third lecture. done fourth, doing second )
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
65 Terms
Describe autosomal recessive disorder
Phenotype does not appear in every generation. Both parents must be carriers (but not affected by the condition). Transmit condition in accordance with Mendelian inheritance.
What is a complex disease?
Are common (66% lifetime risk). Multifactorial (polygenic, environmental). Identified risk alleles suggest susceptibility (are NOT deterministic). Are familial.
State examples of complex diseases that are psychiatric conditions
Alzheimer's, bipolar affected disorder, depression, Tourette’s syndrome.
State examples of complex diseases that are congenital disorders
Cleft lip and palate, neural tube defects, pyloric stenosis.
State examples of complex diseases (that are not psychiatric or congenital)
Obesity, rheumatoid arthritis, insulin dependent diabetes (type I), insulin independent (type II), multiple sclerosis, premature cardio-vascular disease, epilepsy, peptic ulcer, hyperthyroidism, certain cancers.
Why identify genetic components of complex disease?
Early diagnosis and treatment. Allow lifestyle changes to lower risk. Help understand the molecular basis to develop therapeutics.
How many common variants of risk alleles are there? (single nucleotide polymorphisms, SNPs)
11-15 million.
What regions of the genome can SNPs be found in?
Both coding and non-coding.
What is the second most common allele in a population?
Minor allele frequency (MAF).
Identifying alleles which increase risk requires what?
A large number of individuals.
What does family based linkage analysis do?
Method to find risk alleles in complex disease. Looks for linkage between mapping markers and occurrence of disease in families.
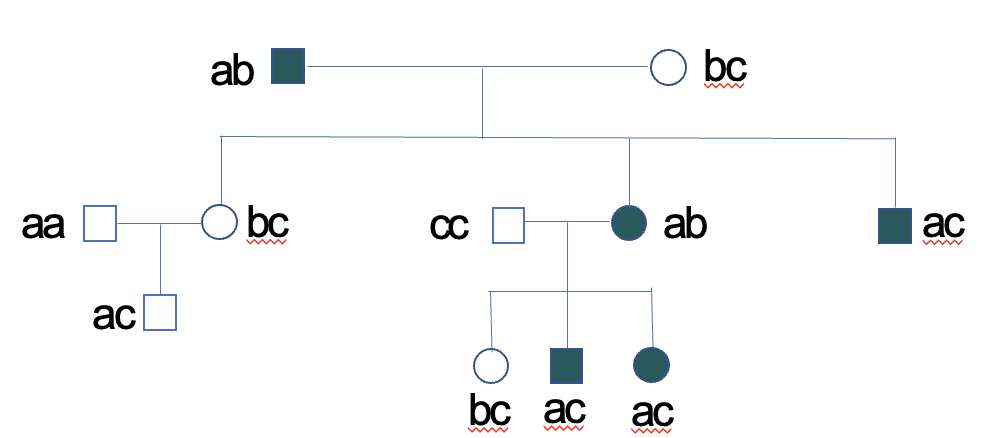
What does genome-wide association studies (GWAS) do?
Method to find risk alleles in complex disease. Search for alleles in a population that occurs more frequently in disease cases then in matched controls. More powerful at identifying rare risk alleles and those that contribute a small increase in risk.
What is phenotypic variation? + give then equation.
Sum of genetic and environmental variation. Vp = Vg + Ve.
What is heritability? + give the equation
The degree of variation within a population that is due to genetic variation. Heritability = Vg/Vp
Why is heritability of a disease not a fixed number?
It can change depending on population assessed and the environment they are in.
What does a higher heritability score mean?
Greater the genetic contribution.
How can heritability be experimentally determined?
Twin studied allows the effect of genetics + environment on phenotypic variance. Monozygotic twins share 100% genes + environment. Dizygotic twins share 50% of alleles + have shared environment. Assume equal environmental influence for identical + non-identical twins.
What is the ACE model for phenotypic variation? + what is the outcome of this in twin studies?
Is what proportion of variance in genetic + environmental.
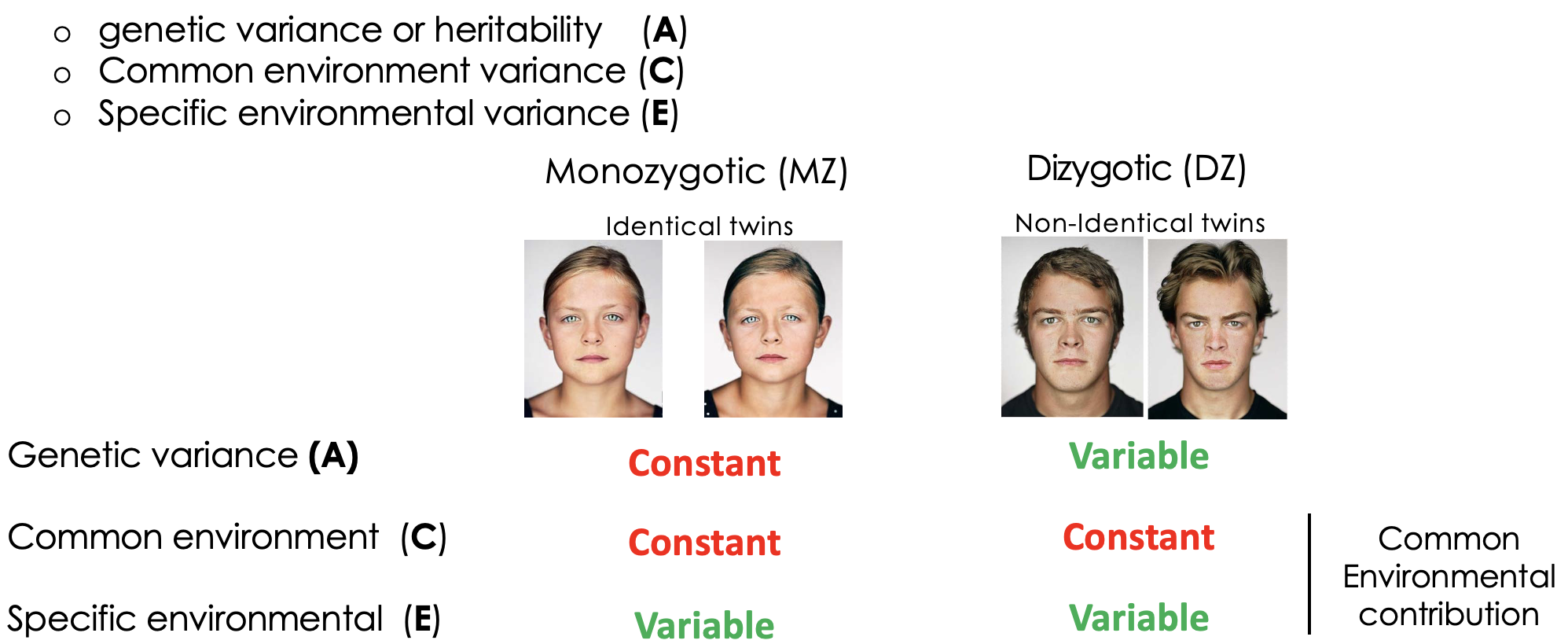
Define concordance in the context of twin studies
Percentages of identical or non-identical twins that share phenotype/disease. Differences is a measure of the effect of 50% of genes on variation in the population. Greater the difference between mono + dizygotic twins the greater the heritability of the trait.
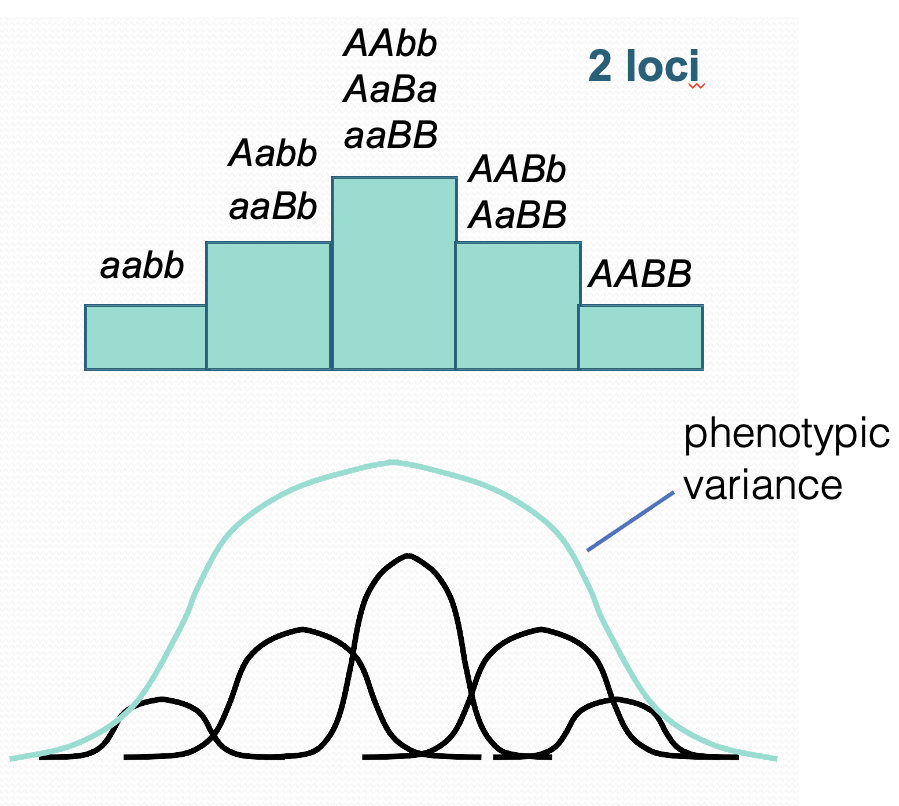
What does it mean if a phenotype shows continuous variation?
Some complex diseases also show a spectrum of variation. Disease is controlled by multiple polygenes. Each allele segregates according to Mendelian laws. Many polygenes + environment = continuous phenotypical variation.
Describe developing disease
Most diseases are discontinuous. An individual either has or does not have the condition.
Describe how polygenic diseases can show a continuous phenotype? **
In complex disease many loci may contribute to this variation leading to broad range of phenotypic variation. Environmental factors will also affect phenotype; the same genotype will display different phenotypes. Not all alleles will contribute the same risk. Increase in the number of loci the greater the distribution of variation.
Describe the threshold hypothesis for disease susceptibility
Most diseases phenotypes are discontinuous. Polygenes + environment produce a distribution of liability. The disease occurs when liability exceeds threshold. Relatives of affected individuals have an increased risk.
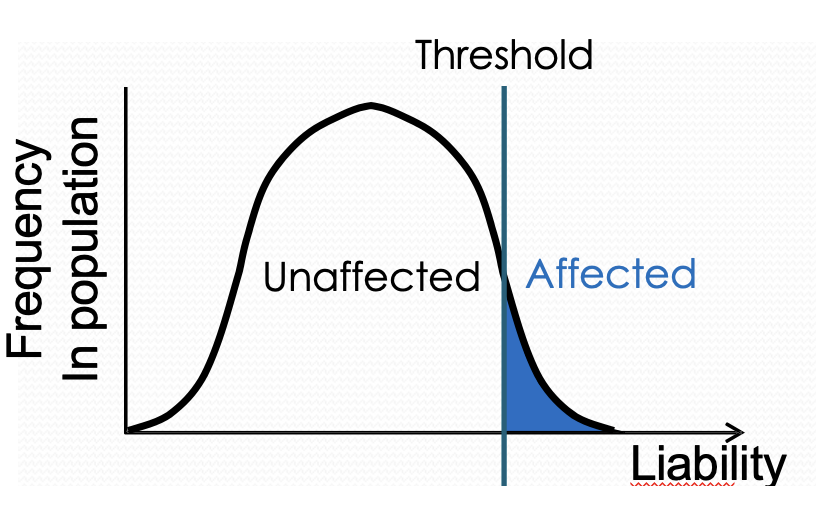
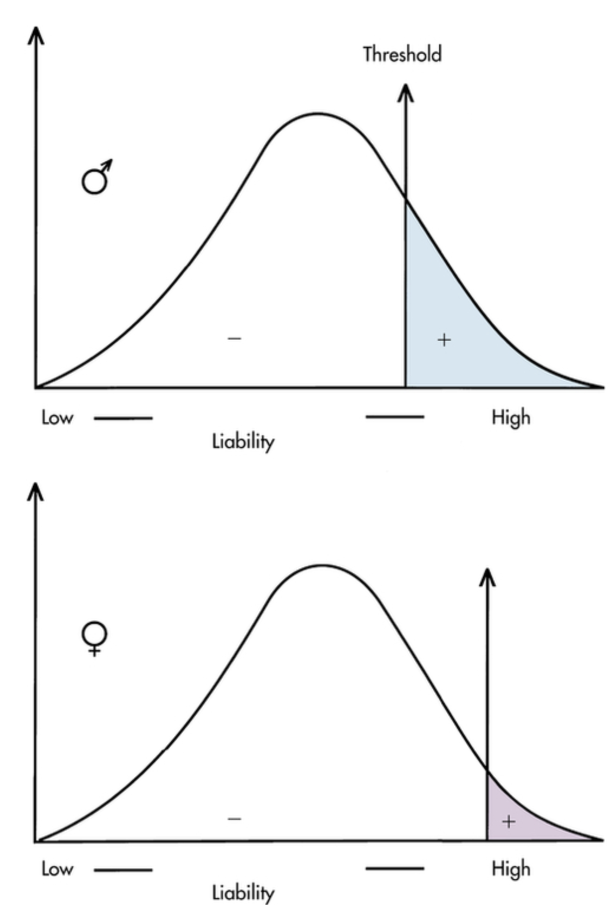
State evidence for threshold hypothesis (pyloric stenosis) **
Narrowing of the opening between stomach + small intestine. Projectile vomiting after feeding in infants. Multiple genetic + environmental factors have been identified as contributing to disease. 5x more common in male infants. Females have a higher liability threshold than males. Affected females carry more risk alleles. Relatives of affected females have greater risk than affected males.
State the different possible architectures of complex diseases (parkinson’s, type II diabetes, breast cancer)
Parkinson’s: small number of dominant alleles conger a large increase in risk.
Type II diabetes: common disease, common variant model (CDCV) - many alleles confer a small increase in risk.
Breast cancer: intermediate- one major allele exerts a large effect, numerous other lower risk alleles.
State different possible architectures of complex diseases **
Small number of dominant alleles confer a large increase in risk. Common disease, common variant model - many alleles confer a small increase in risk. Intermediate - one major allele exerts a large effect, numerous other lower risk alleles.
Describe single nucleotide polymorphisms (SNPs)
Between 11 and 15 million common SNPs. Uneven distribution of SNPs in the genome. Can occur at coding regions (synonymous or non-synonymous) or non-coding regions (can affect expression/regulation of associated genes). Complex diseases arise from combinations of multiple SNPs.
Define synonymous
No change in encoded a.a.
Define non-synonymous
e.g missense or nonsense mutations.
Describe genome and exome sequencing
WGS: sequence both coding + non-coding regions.
WES: sequencing only protein encoding regions of genome.
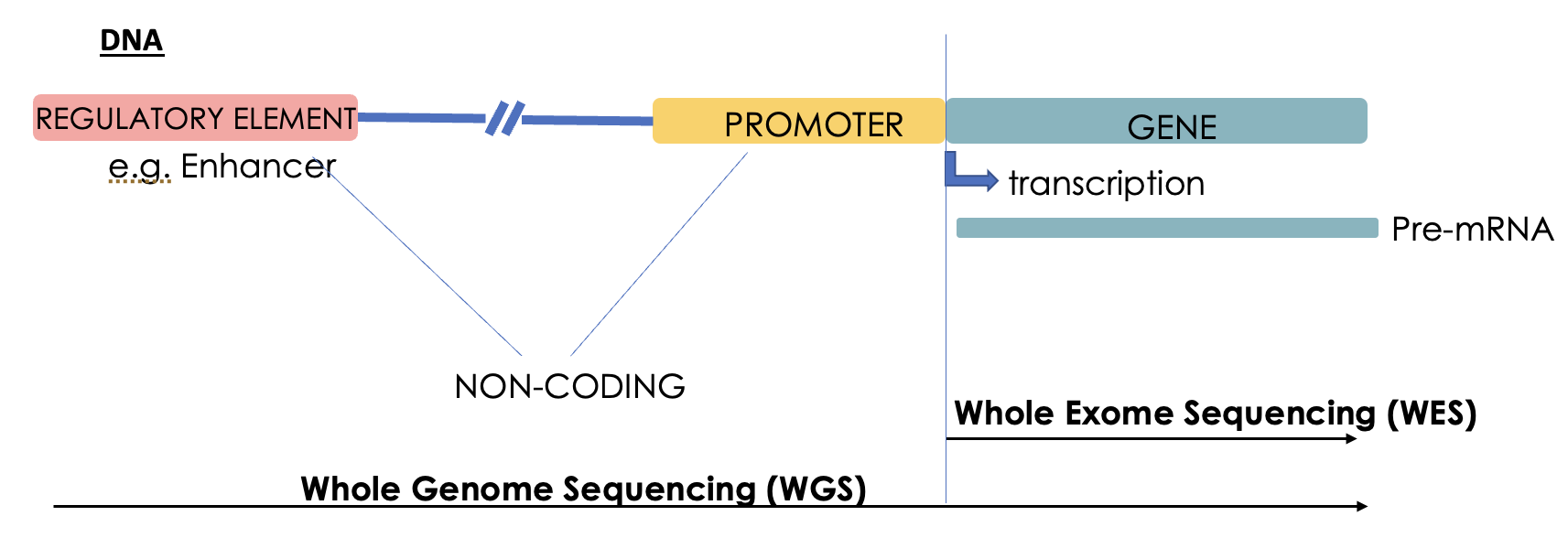
Describe gnomAD database
Exome + genomes from unrelated individuals sequenced as part of various disease-specific + population genetic studied. Aggregates over 730,000 exome + 76,000 genome data sets. Over 910 million variants mapped. Records frequency of alleles in a population = documents rare mutations.
Highly pathogenic variants are seen with a what frequency in the general population?
Lower.
Describe Genome Wide Association Studies
Examine a panel of SNPs in the genome for association with the disease phenotype. Compare the allelic frequency across the entire genome in case + control population. Needs many participants. Have been done for most common diseases. Many risk loci yet to be identified. Missing loci contribute to the “missing heritability problem”. Sig diffs in allelic frequency constitutes an association with disease.
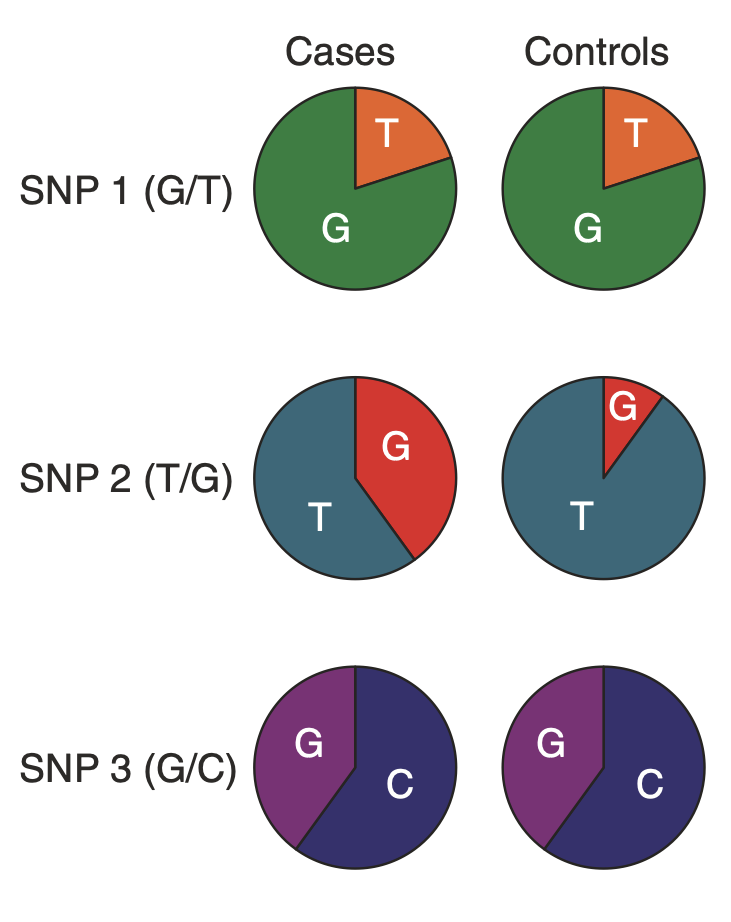
How are SNPs associated with disease?
SNP itself can increase risk. The SNP correlates with the risk allele due to linkage disequilibrium.
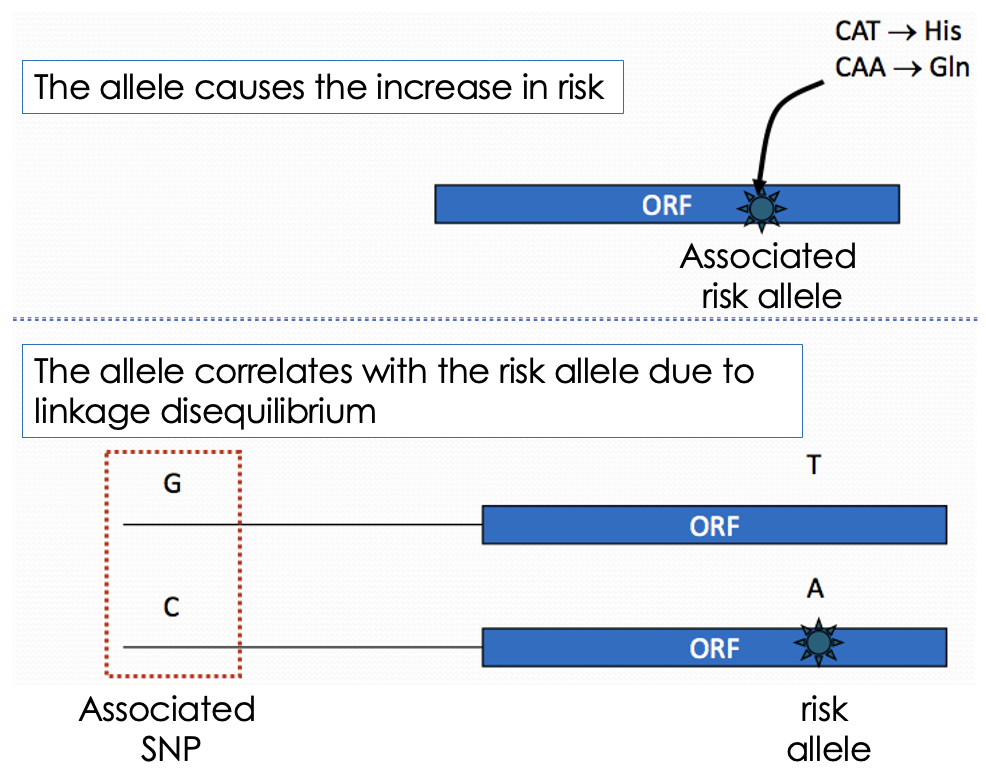
Define linkage disequilibrium (LD)
Is the non-random association of alleles at different genomic sites.
What does linkage disequilibrium (LD) depend on?
Distance between alleles (closer together tend to be inherited together). Recombination rate.
What can patterns of linkage disequilibrium in the human genome be summarised as?
Haplotype blocks. Are regions of high LD that are separated from other haplotype blocks by many historical recombination events. In haplotype mappings, groups of alleles are clustered so a single SNP can identify the cluster of alleles (tag SNP).
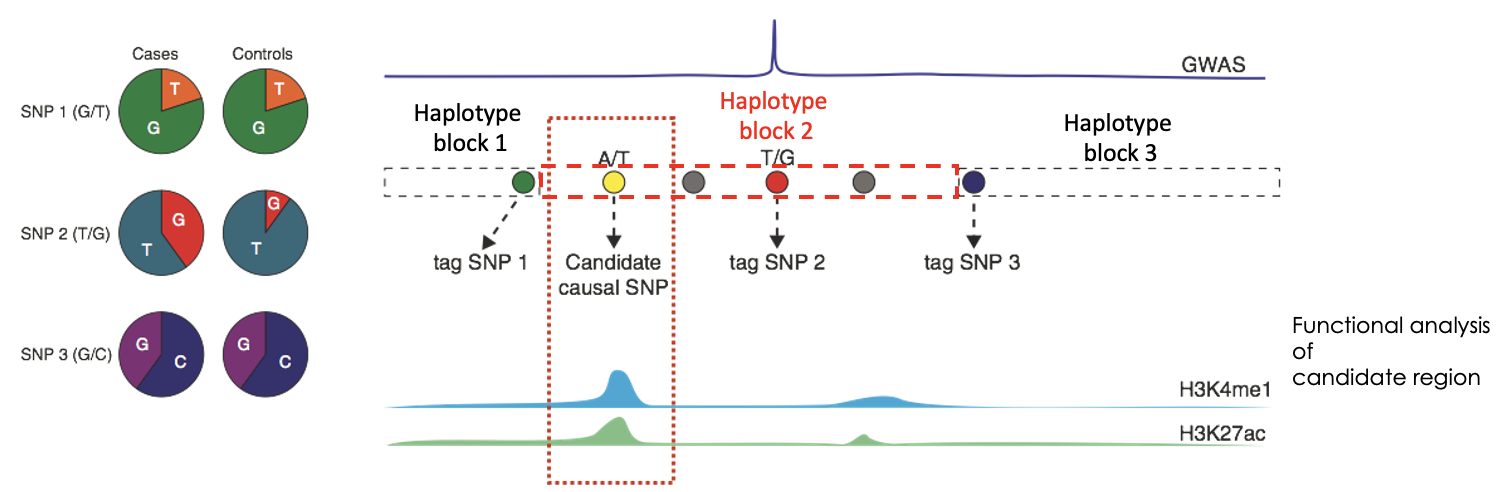
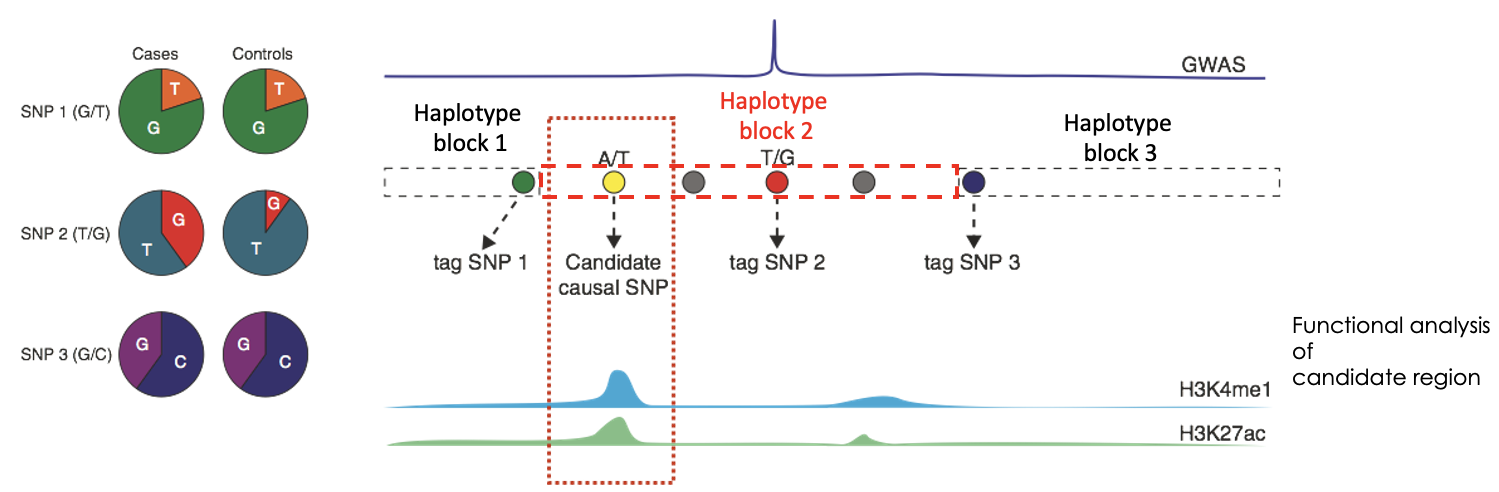
What do you need to do after identifying the which SNP is linked to which haplotype block?
Integration with functional data on candidate regions to identify causality. Can use GWAS studies to do this.
What is used to measure the likelihood of a SNP being associated with a disease?
Odds ratio (OR).
Define odds ratio (OR)
Statistic that quantifies the strength of the association between two events.
OR = 1 (Events are independent)
OR > 1 (Events are correlated)
OR < 1 (Events are negatively correlated)
What is the common disease common variant (CDCV) model of complex diseases?
Multiples alleles with OR < 1.2 showing weak association to the disease phenotype.
What does GWAS rely on?
Statistical analysis + large cohorts. Statistical significance is needed to differentiate true +ives from false +ives.
What is GWAS susceptible to?
High number of false -ives.
Describe type II diabetes
Common chronic condition caused by inability to take up sugar. Characterised by high blood sugar, insulin resistance + lack of insulin production. Is multifactorial: familial, geography + ethnicity, age, weight diet + level of physical activity.
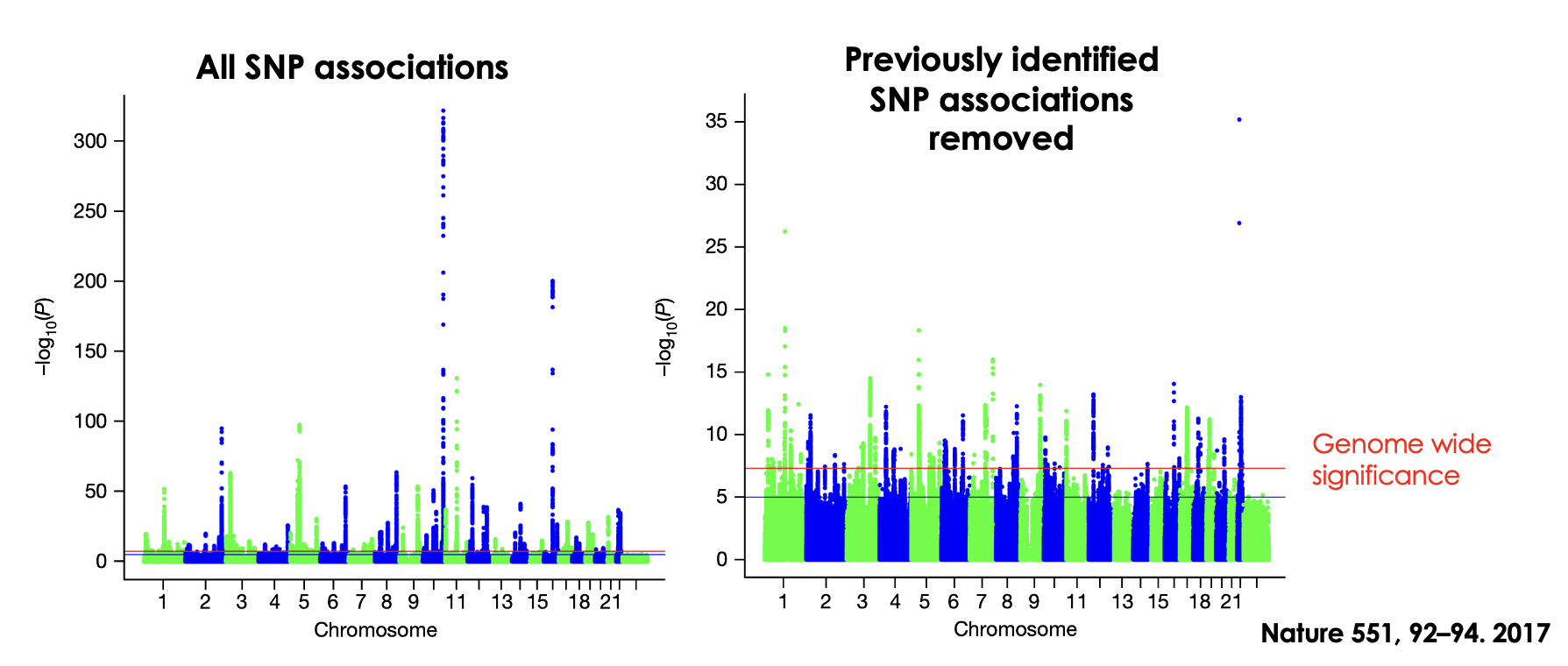
Outline GWAs of type II diabetes
Previously identified common risk alleles (red SNPs). Novel association loci determined due to increased statistical power (green SNPs). These novel loci have low Odds ratio (1.06-1.27) with each causing only a small increase in risk.
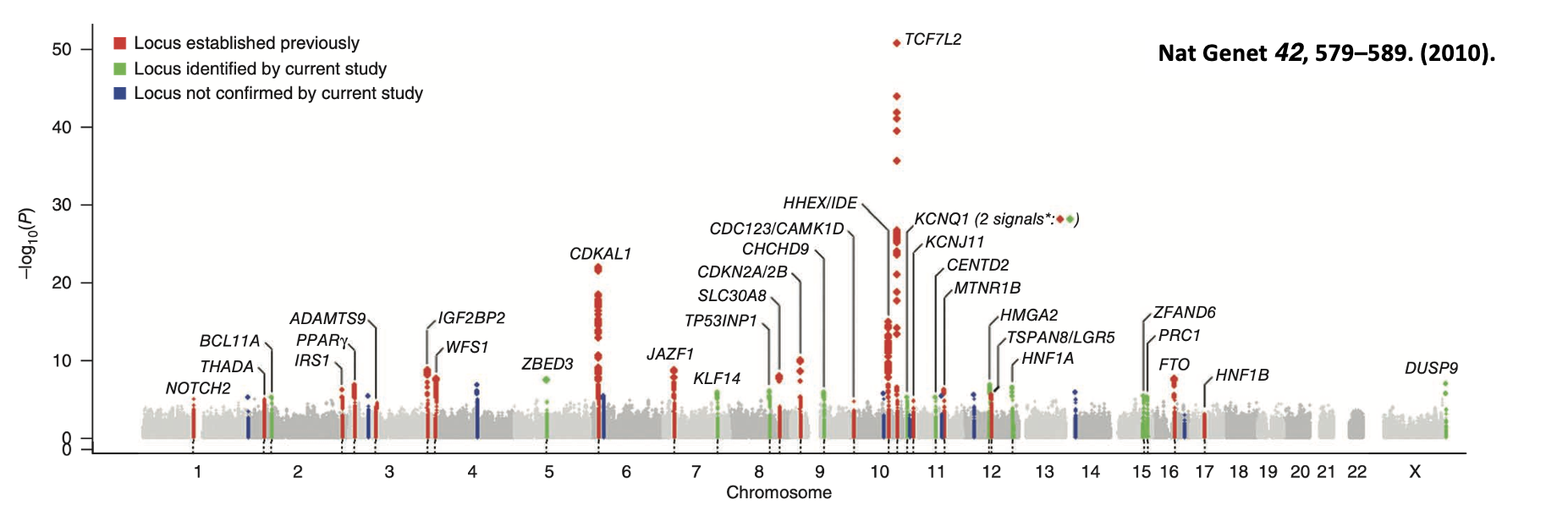
In relation to type II diabetes, briefly describe the following alleles: TCF7L2, FTO, CDKN2A/B
TCF7L2: allele providing the greatest risk of type II diabetes. Intronic variant. TF required for pancreatic development.
FTO: intronic variant. Involved in body weight regulation.
CDKN2A/B: non-coding regulatory variant.
Describe breast cancer
Lifetime risk is 8-12% in females. The risk of disease increases if first degree relatives suffer from condition. Rare coding mutations + common non-coding variants increase risk of disease. Rare coding mutations (e.g BRCA1 + BRCA2) greatly increase risk. Common variants in polygenes contribute small increase in risk. 5% of cases have BRCA1/BRCA2 autosomal dominant alleles. BRCA1 + BRCA2 were mapped by linkage analysis.
Outline GWAS of breast cancer
Identify high known high-risk factors that occur infrequently in the population. Identify 66 common low-risk alleles many of which are in the non-coding region of the genome.
Define heritability
Proportion of variance in a particular phenotype in a population that is due to genetic variation. Can identifiy risk alleles for complex disease but heritability is not fully explained by the alleles.
What can missing risk arise from?
False -ives in GWAS studies. Rare variant alleles with a MAF 1-5%. Structural alteration of the genome. Epigenetics. 3D genome organisation.
Single gene disorders are what?
Individually rare but collectively common.
State 2 autosomal diseases
Huntingtons disease (autosomal dominant). Cystic Fibrosis (autosomal recessive).
State an X-chromosome linked disease
Duschenes Muscular Dystrophy.
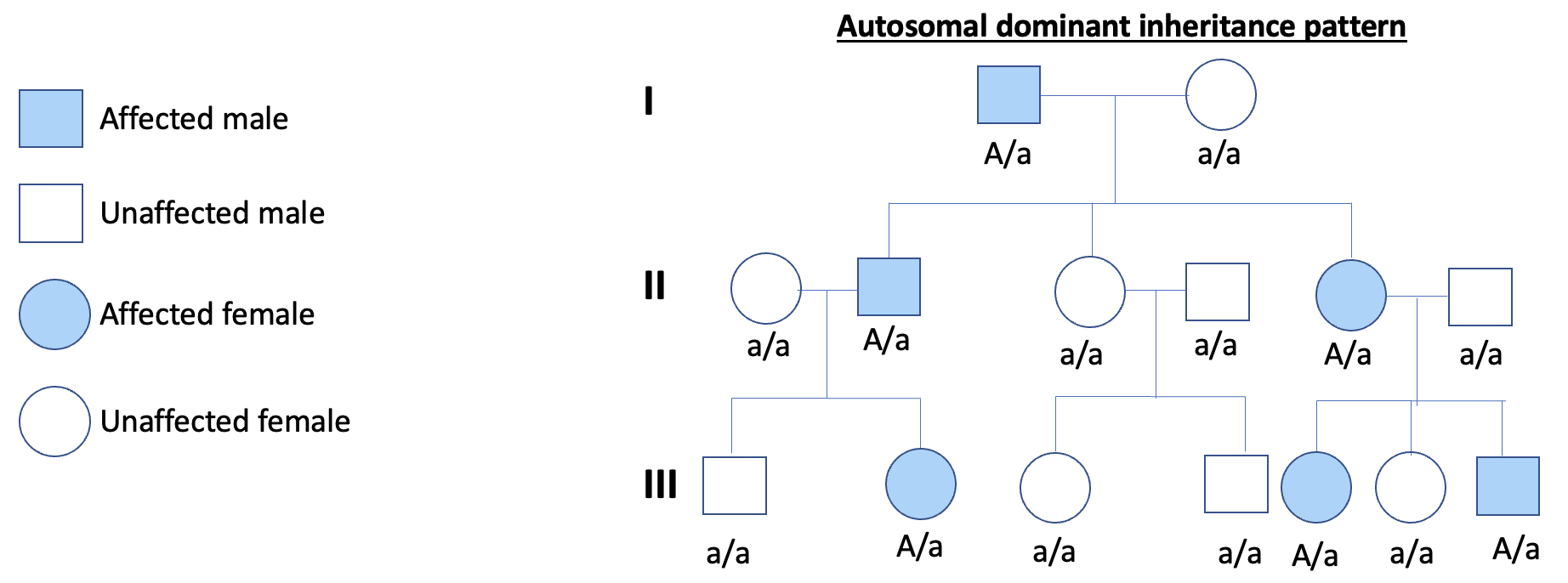
What is an autosomal dominant disorder?
Where 1 copy of mutation is sufficient for the individual to be affected. WT allele is recessive + the defective alleles is dominant. More likely that one copy of the defective allele is present than two. Inherited in a dominant pattern OR a new mutation arises in a single copy of the gene. Phenotype appears in every gen.
Why are the typical mendelian inheritance ratios not seen in humans when looking at autosomal dominant disorders?
Small progeny sample size.
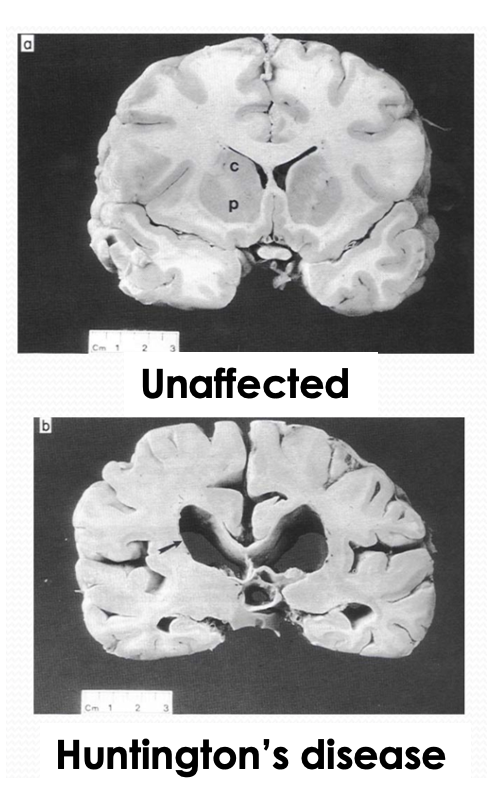
Describe Huntington’s disease
A late onset autosomal dominant disorder. Affects 1 in 10,000. Is a progressive neurodegenerative brain disorder that causes: neuronal death, cerebral atrophy, CNS disorder, uncontrolled movements, decrease in cognitive function, personality changes.