Biochemistry Exam 2
1/122
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
123 Terms
trypsin
endopeptidase
serine protease = recog Rn-1 (amino that contrib carbonyl C)
most specific
recog pos charged residues: Arg, Lys
does not work w Pro (induces kink so cannot cleave)
chymotrypsin
endopeptidase
serine protease = recog Rn-1 (amino that contrib carbonyl C)
recog bulky hydrophob residues: Phe, Tryp, Tyr (aromatic side chain)
does not work for Pro
pepsin
endopeptidase
aspartic protease = recog Rn (amino that contrib NH))
unspecific: Leu, Phe, Trp, Tyr
does not work with Pro
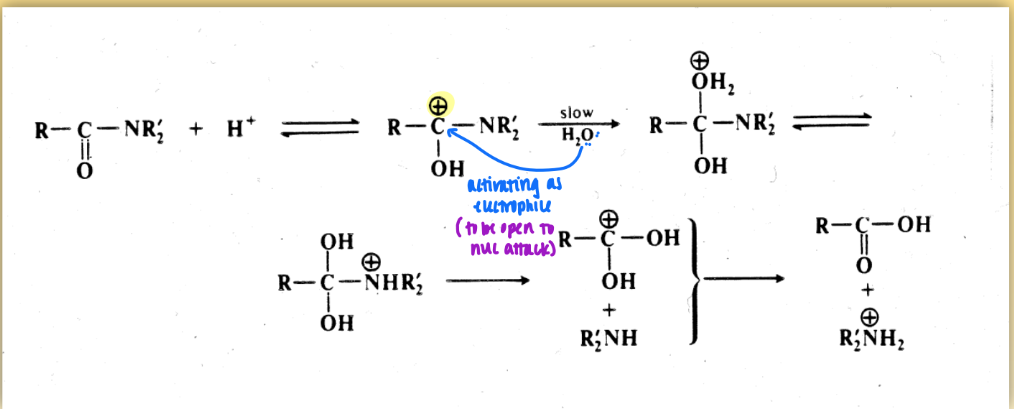
amide bond hydrolysis: acid mediated mechanism
for aspartic protease
not catalyzed event = reactant not regenerated
includes electrophile & nuc attack
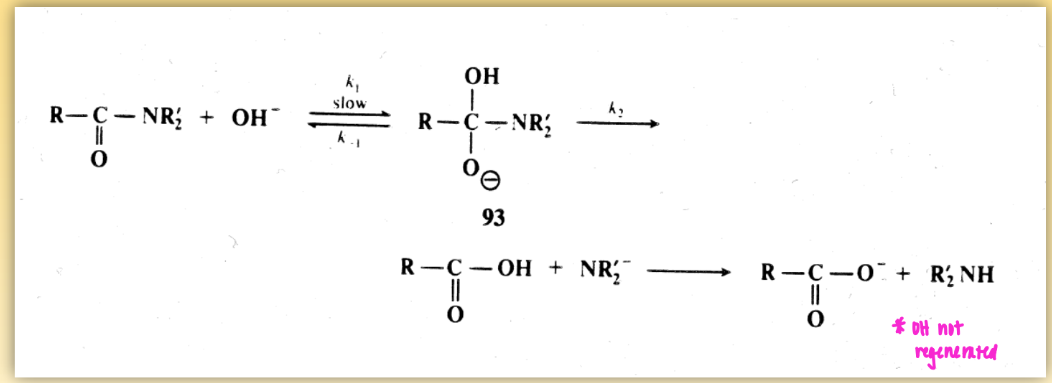
amide bond hydrolysis: base mediated mech
for serine proteases
OH not regenerated
catalytic triad
Asp - His - Ser
always conserved in active site of enzyme of serine protease
uses low barrier H to enhance nucleophilicity/ base characteristics → leads to easier breakdown via nuc attack
acyl-enzyme intermediate = molec cov modified
cynanogen bromide (CNBr)
cleaves peptide bond (internally)
recog Met (high specificity)
analogous to serine protease (endo)
end w/ modified residue: homoserine
C-term not free
unlike ser prot that no mess w/ polypep chem
determining primary struc of prot
reduction of disulfide bonds to sep polypep chain
oxidation of disulfide bonds w/ performic acid = irreversible w/ no cap
break down polypep chain w/ two dif methods to gen dif set of peptide frag
ex. trypsin + CNBr (both v spec)
determine seq of each frag w/ edman degradation
use overlapping seq in each pep frag to determine seq of each polypep
repeat frag w/o breaking disulfide bonds so can detect Cys
edman degradation
used to sequence oligopep frags
acid hydrolysis of PTC polypep yields PTH amino acid & intact polypep minus 1 amino acid from N-term
one by one cleave from n-term and seq amino
tag n-term w/ dansyl
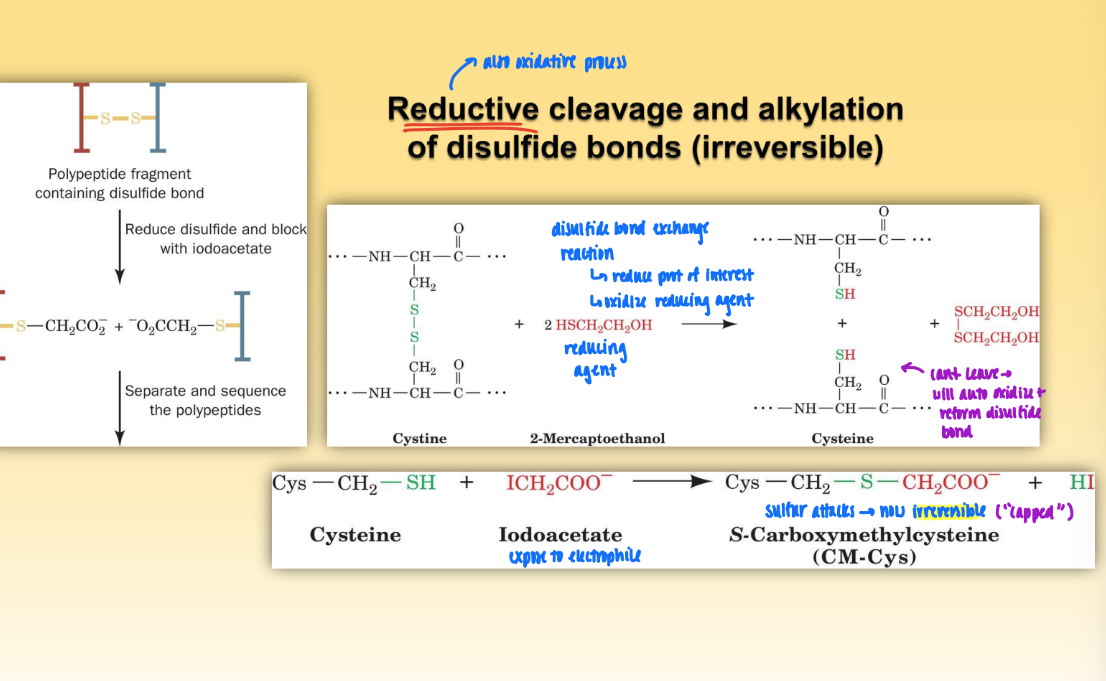
reductive cleavage and alkylation of disulfide bonds
oxidative process = reduce prot of interest & oxidize reducing agent
expose to electrophile (iodoacetate) & sulfur attacks to prevent auto oxidation and reformation of disulfide bonds
this leads to capping of S
merrifield synthesis process
use boc group to protect n-term & protecting group to protect side chain of amino acid of interest
couple resin w/ fxnal group to boc+aa+protecting
connects desired aa to resin via free COOH group
remove boc group to free amino group
attach second aa w/ free COOH (but has boc group to protect n-term)
continue w/ desired number of aa
remove protecting groups & then resin via chromatography
= mature oligopeptide
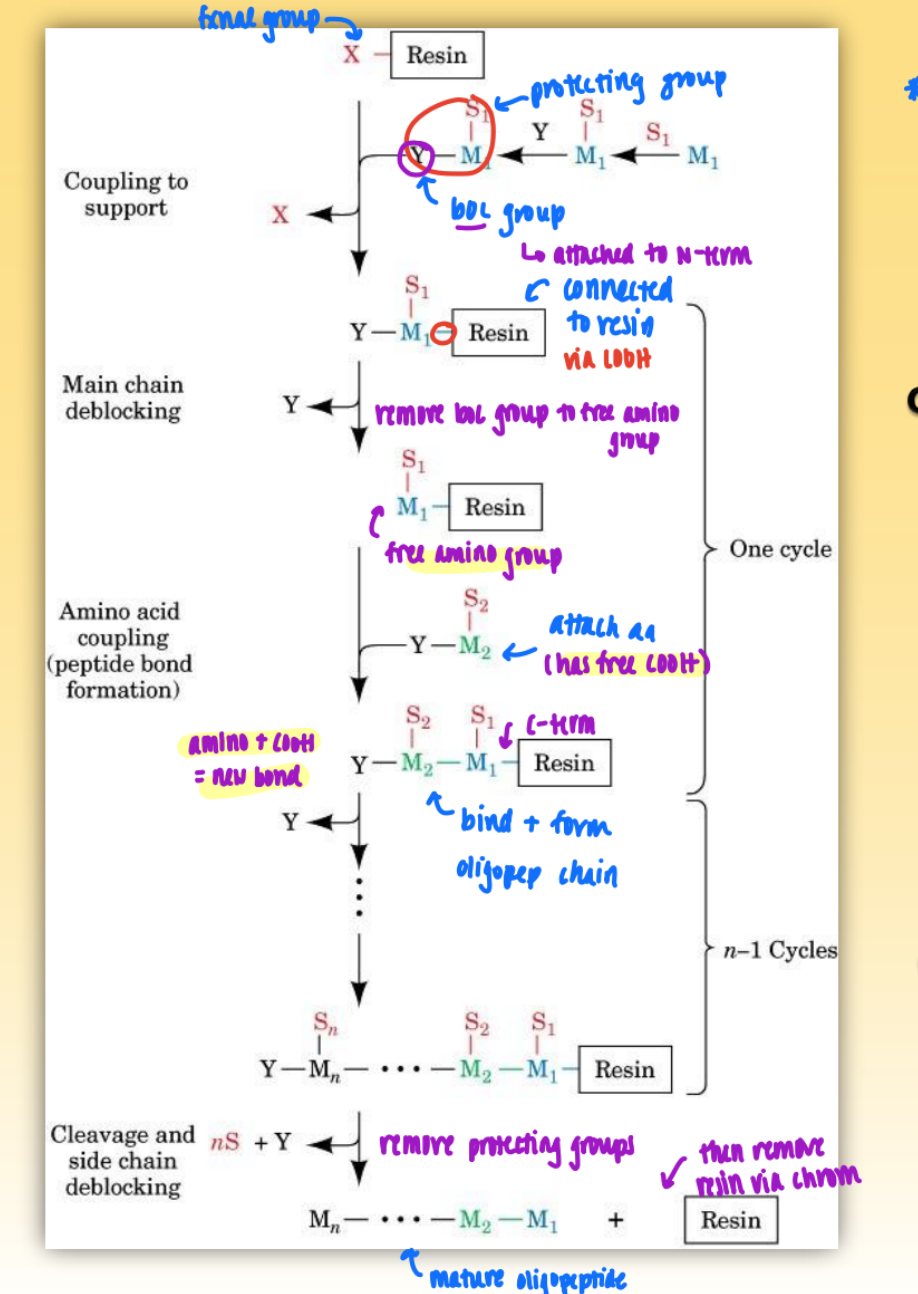
merrifield synthesis overview
synthesize c-term to n-term
used to chemically synthesize polypep in solid phase (on column)
want to install aa on resin
ensure go to completion!
if low yield = nested frag = hard to purify
limitations: 100-150 aa
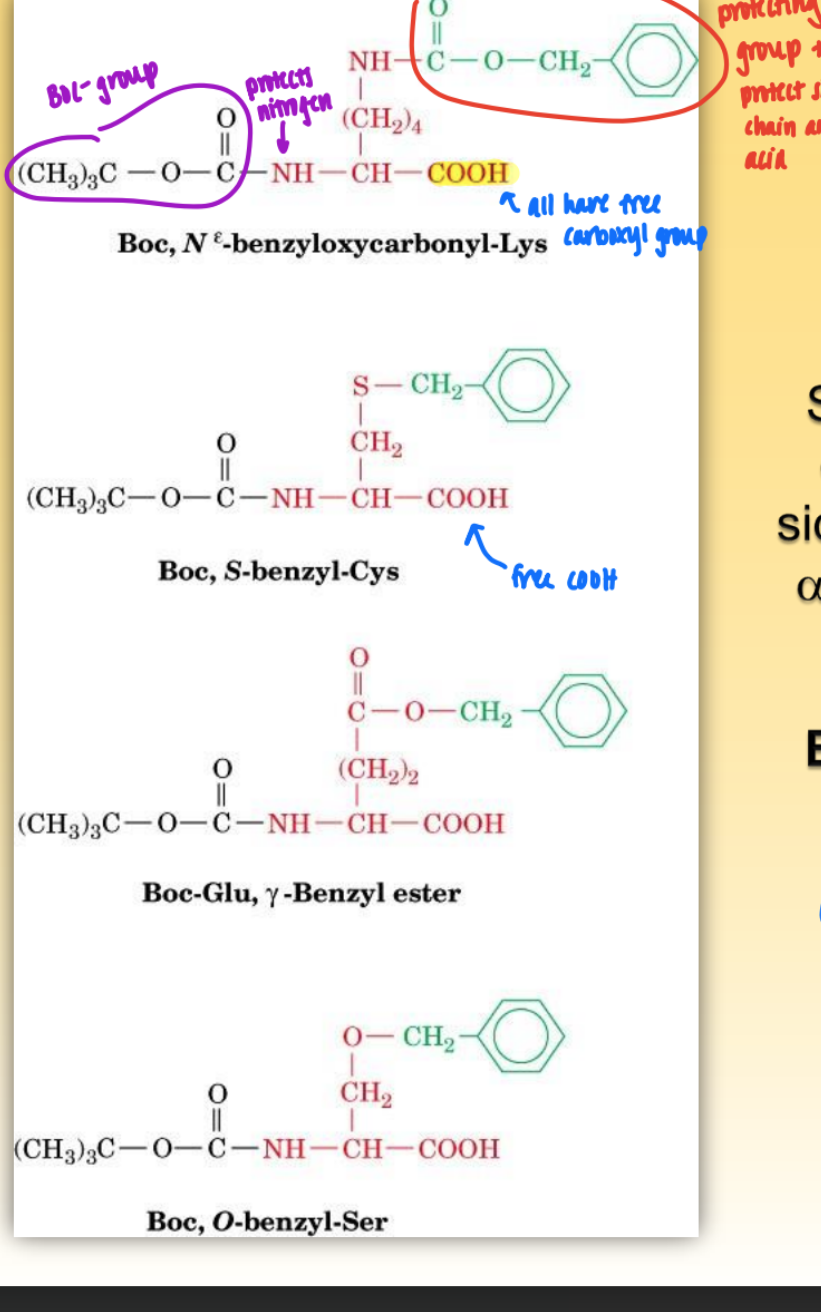
boc group
= protecting group on side chains w/ specificity
protects amino group
used in solid phase peptide synth (merrifield)
all have free COOH
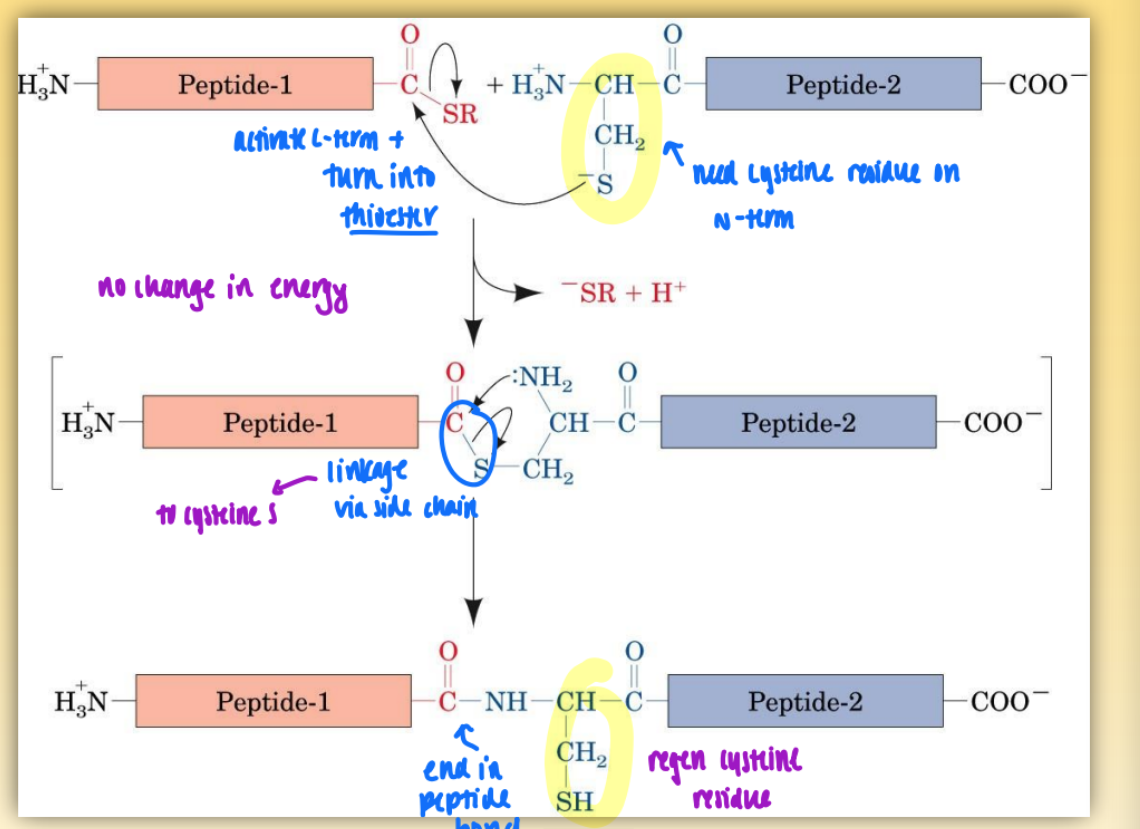
linkage of 2 peptides chemically = native chem ligation
activate c-term & turn into thioester (using cysteine residue on n-term)
link via side chain to cysteine S
end in peptide bond & regen cysteine residue
two peptides could be made via merrifield
first step = no change in energy
second step = favorable
mass spectrometry (MALDI)
MALDI = matrix-assisted laser desorption/ionization
mass usually = molec weight = vap/ionize molec = too strong & can break polypep chain
MALDI = get molec weight of polypep but gentle = keep prot intact during vap/ionize
can determine accuracy of synth prot if mass same as expected (count up aminos)
conjugated protein
prot prodiced via cov interactions
ex. glycoprot, phosphoprot, hemoprot
analytical purification
small prot applied to small, compact column (ug, ng)
preparative purification
bench work = big prot (mg, g) = purify a lot of prot
salting out (NH4)2SO4 precipitation
continuously increase concen of (NH4)2SO4 to keep precip out new prot
crude = does not lead to homogenous prot → but get as purified as possible & cut down contaminating prot
membrane dialysis
used to sep by molec weight = small and large molec
use dif pore sizes w/ cutoffs (keep desired prot in bag and everything else in solution)
used to desalt prot (use very small prot)
crude = not completely pure
osmotic pressure
water wants to goes back into bag so concen of big molecs = same
(why we need to seal bag in membrane dialysis)
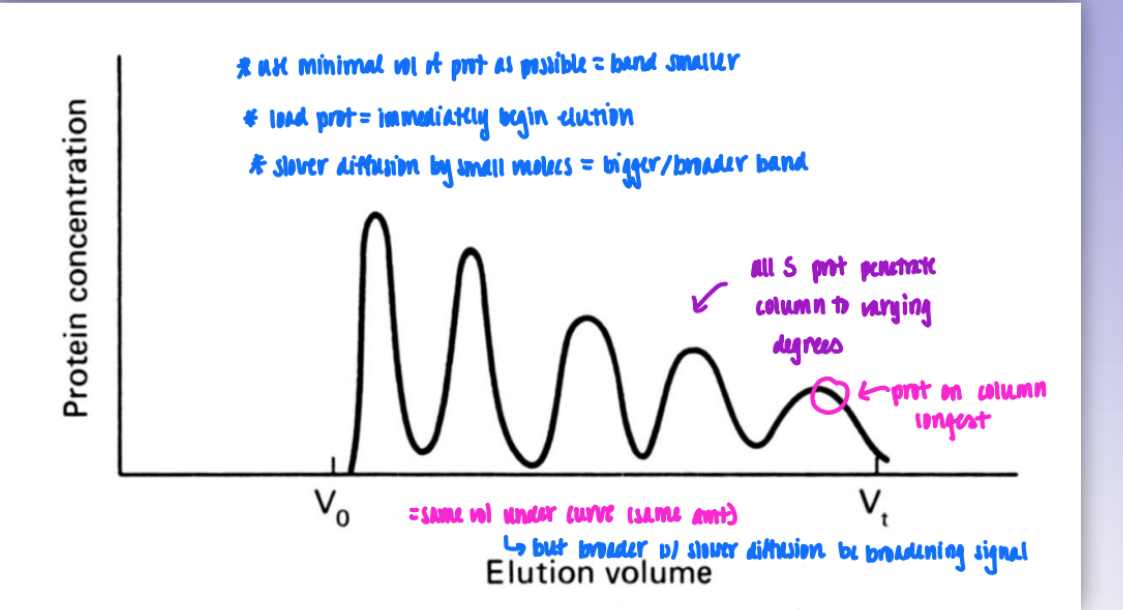
gel filtration / size-exclusion chromatography
sep of prot based on dif in size (and secondary dep on shape)
use size-exclusion resin w/ dif porosities (large v small holes)
pack into column & load mixture
small molecs can penetrate beads = small elute slow
large molecs = excluded from column = big elute fast
residence time & band width
as prot size decreases = elutes slower = on coulmn for longer = last peak
small molecs = broader band = slower & greater diffusion
but same vol so area under curve of each peak = same
exclusion limit
molecular mass of the smallest molecule unable to penetrate the pores (beads) of given gel
Vo
void volume
volume of solvent space surrounding the gel beads = determined experimentally by using large standard molec like blue dextran
Vx
volume occupied by gel beads
Vt
total bed vol (= Vx + Vo)
Ve
elution volume = volume of solvent required to elute a solute from the column
Ve/Vo
relative elution volume = indep of the size of the particular column used (used to compare elution behaviors)
SEC Ve/Vo plot
plotting Ve/Vo against logM = linear standard curve
use to determine mass of unknown prot
exceptions due to shape (assume globular nature)
ex. fibrinogen = behaves like larger molec than actual size
ion-exchange chromatography
uses stepwise elution
cation exchange
resin: neg charged, mobile cations as exchangers
large net pos = tightly bind to column
large neg = cannot interact w/ resin → elute fast (“salt out”)
ideal method = usually start so all have net pos charge at beginning of elution & then charge pI for gradual elution in order of increasing pI
ion-exchange resins
can have both ion-exchange & gel filtration habits
size also matters in some cases
size factors can lead to deviations in expected & predicted elution based on pI
elution profile
can predict elution based on pI but size factors can lead to deviations;’l;l
affinity chromatography
most powerful method of purification
effective resin must be used = hardest part
build column by self = extra specificity towards desired protein
only desired prot binds ligand (after washing column)
other contaminants are eluted out
effective resin production
covalently link ligand (amino group) to agarose gel using CNBr as an activator
activates OH group in agarose gel
amino group = spec to desired prot
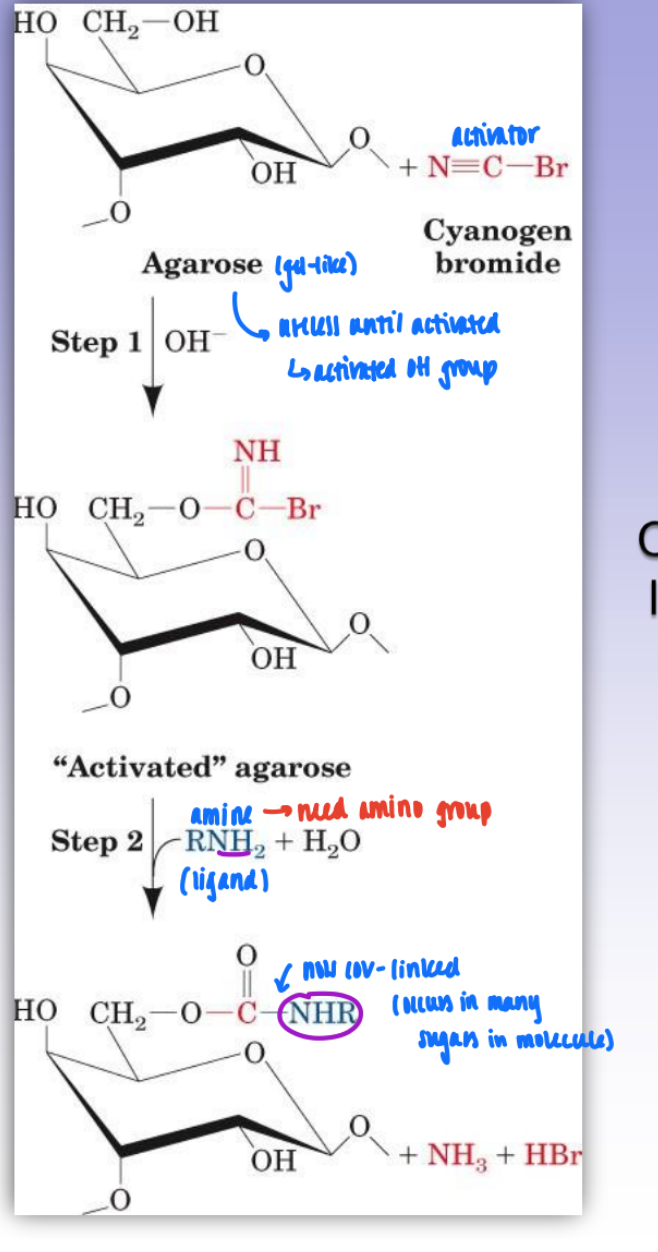
derivatization of epoxy-activated agarose
gel pre-activated as epoxide
makes it easier to link & more reversible
link epoxide & ligand
spacer arm = dist between struc of ag bead & functional group
sterics important
if ligand too close to bead = sterically inaccessible & binding will not occur
purification of staphylococcal nuclease by affinity chromatography
on agarose
gel binds to nucleotides like amino acids
resin is built w/ T-residue
modified t-ligand = so amino group is attached & can bind resin surface
leads to very homogenous mixture
wash out excess prot = only desired prot bound
able to access bound prot
after washing = elute w/ actual ligand = easily and cleanly remove protein
slab gel electrophoresis
separation (rate of migration) based on differences in mass/charge ratios of proteins
prot migrate from cathode to anode
pH 9 buffer used
formation of cross-linked polyacrylamide gel for electrophoresis (PAGE)
polymerization of acrylamide & N,N-methylenebisacrylamide
use radical polymerization (not very structured)
can change ratio of acryl & N,N → changes porosity
thus changes % of cross-link
Ferguson plots
log mobility over % gel
free mobility at 0% gel
when size does not matter (only dependent on charge)
as % gel increases = size matters
lose mobility
small particle, high charge = high mobility
big particle, low charge = low mobility (greatly impeded)
SDS-PAGE
SDS = amphipathic
SDS-treated proteins = identical mass:charge ratios
separation is solely based on molecular mass
can allow us to estimate molec mass of unidentified prot using standards
relationship between motility & mass is logarithmic
SDS-PAGE & multi-subunit proteins
gives molec mass of prot subunits, not whole prot
each polypep chain will have own band
= disrupts the non-cov bonds that hold quat strucs tg
ultracentrifugation
sedimentation coefficients for some bio materials
vary from small (small prot) to big (mitochondria)
fractionate subcellular organelle
based on mass and shape secondarly
zonal ultracentrifugation
uses preformed sucrose density gradient
then add sample & spin at high speed (centrifugation)
let settle & see fractionation (particles sediment)
slow v fast sedimenting components
can then poke holes and sep each fraction
use sedimentation coefficients to separate (mass based)
okay for proteins!!!!
isopycnic ultracentrifugation
sep mixtures according to density
only involves fractionation of subcellular organelles
prot have same densities
no proteins only organelles
initially homogenous mixture becomes gradient based in density (in relation to density of CsCl or Cs2SO4 solution)
isoelectric focusing
create pH gradient using ampholyte soln incorporated in gel
pH gradient established (neg→pos)
protein added & migrate in column based on pI valued
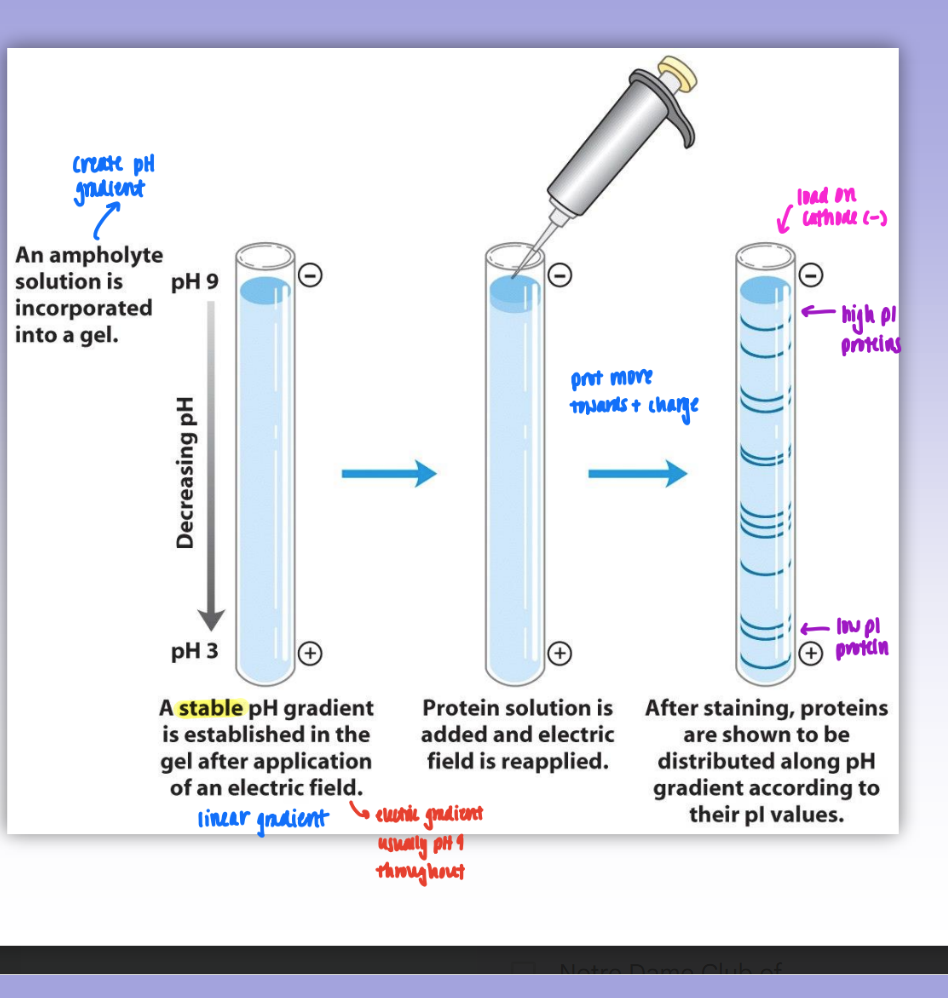
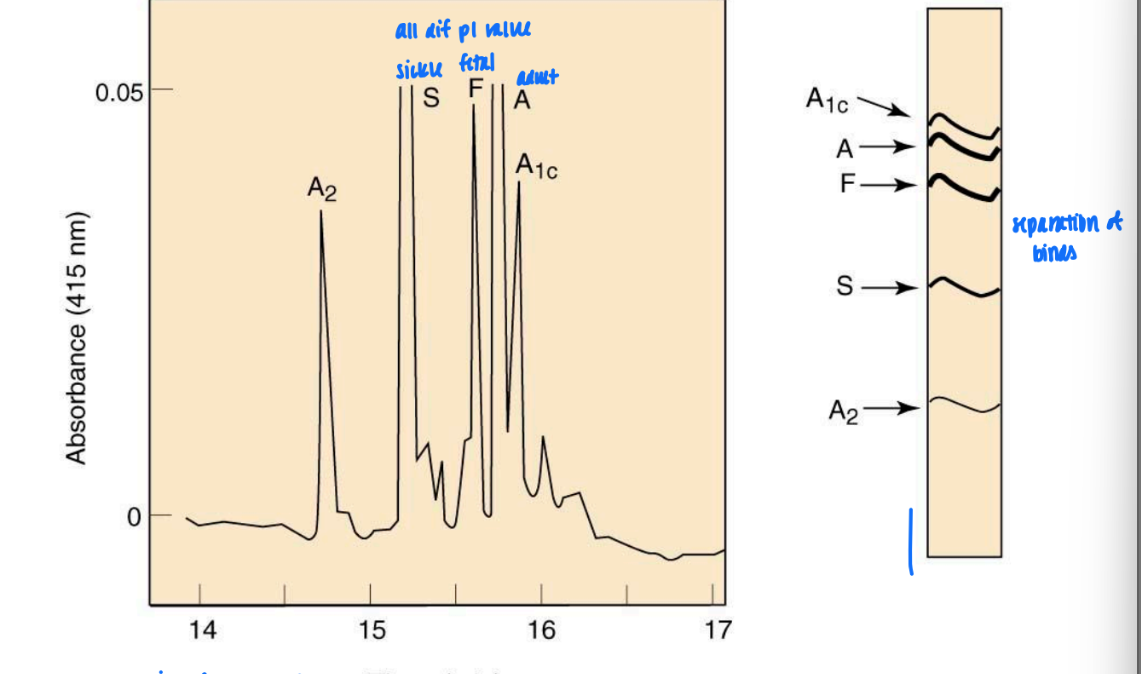
isoelectric focusing of hemoglobins
dif subunits of prot have dif absorbance
dif pI = dif fractionation & good separation of bands
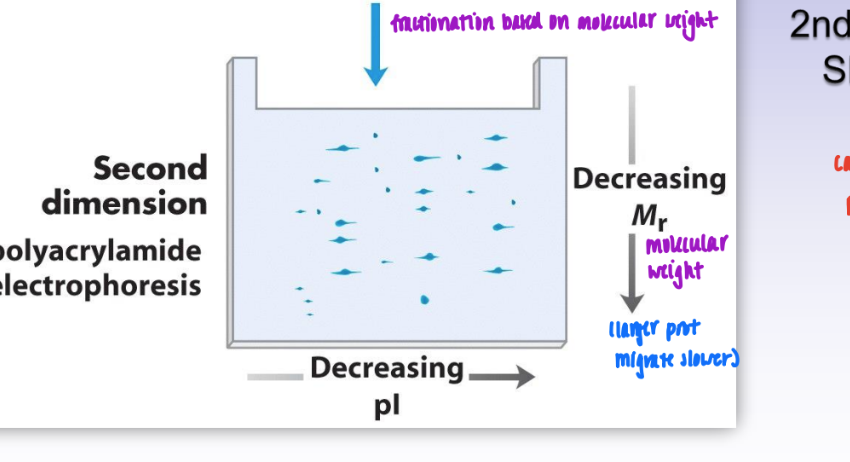
2-dimensional gel-eletrophoresis
1st dimension = isoelectric focusing
fractionation based on pI
prot fall in order of decreasing pI
2nd dimension = SDS-PAGE
based on molec weight
= leads to much better & more complex sep of proteins
peptide bond UV absorption
aromatic absorption at 280 nm
all prot absorb around 200 nm
due to amide fxnal group
not all prot behave similar in amide region of UV absorption
amide absorption bands are dif
dep on secondary bond to amide
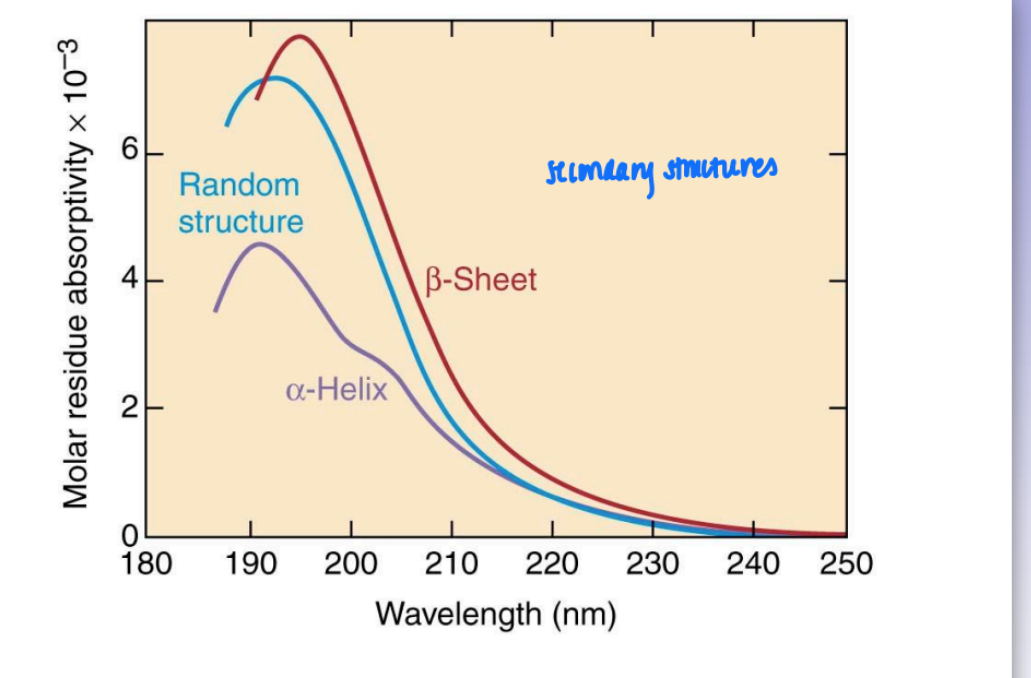
CD (circular dichroism) spectra of polypep
delta E = dif in circular polarized light (measure of chiral props exclusively)
uses polarized light = can distinguish between random coil, alpha, and beta
3 distinctly dif curves (standards)
can use to estimate % of each 2nd struc in protein
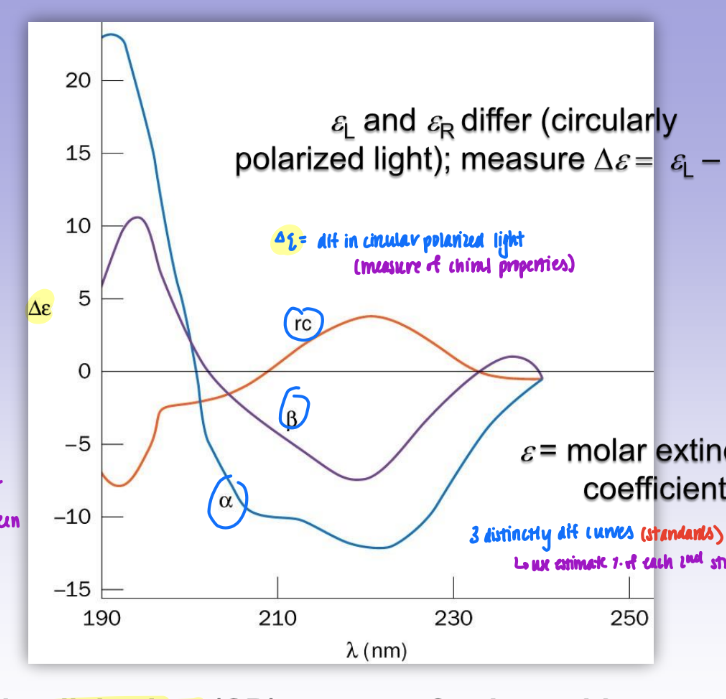
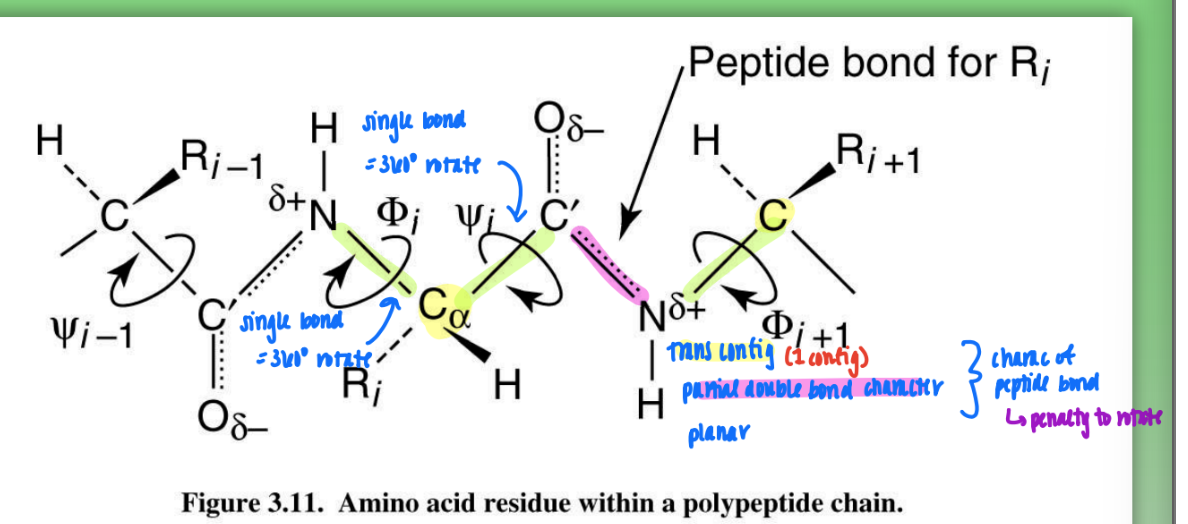
trans peptide (amide) configuration
all atoms lie in same plane
due to geometry around C+N (120 degree bond angles)
C alpha and C alpha 180 from one another
more stable
favored by bigger R groups = less steric hindrance
so most prot trans
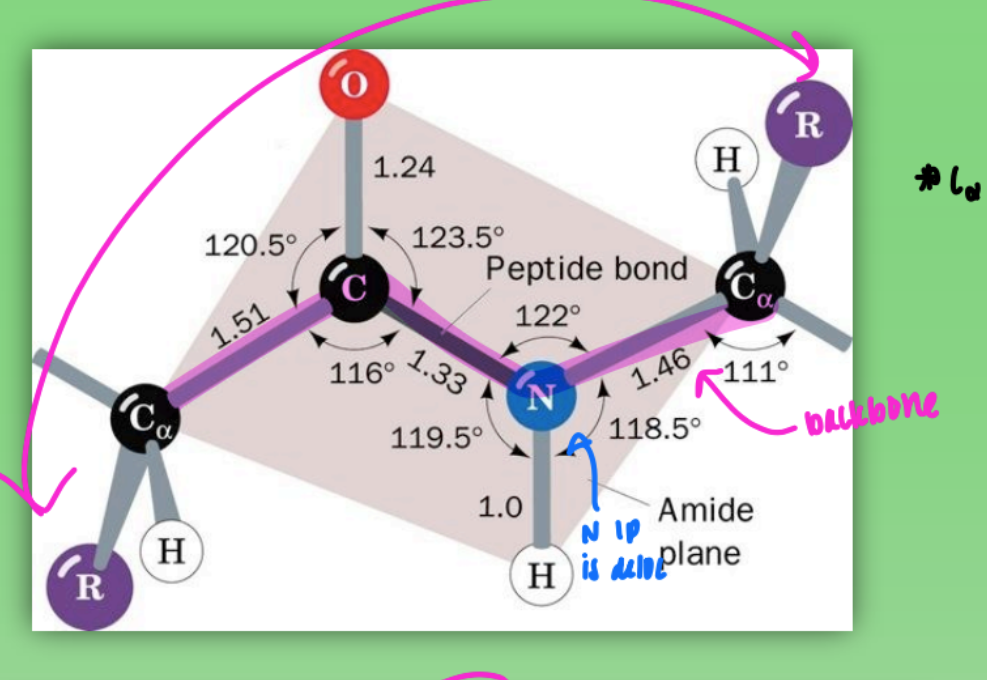
cis peptide (amide) configuration
C alpha and C alpha closer tg
hindered by R groups
C+N still 120 degree
less used by prot except glycine (H)
less stable
when many Gly = mixture of trans & cis = unstable protein
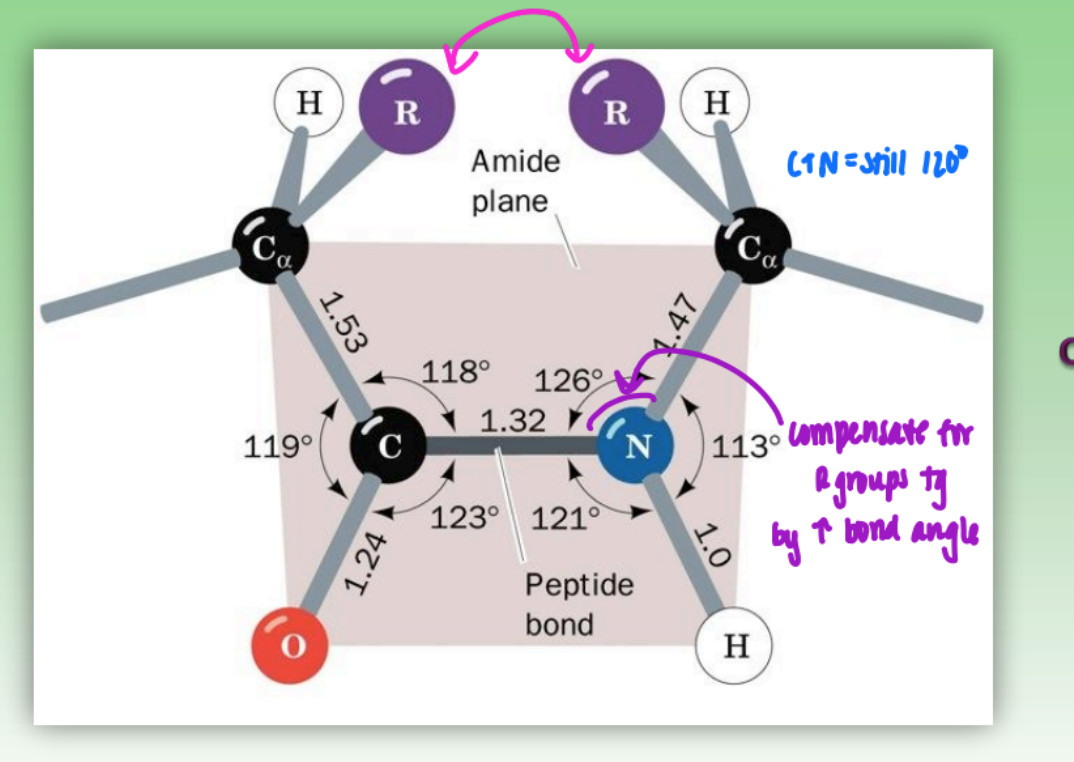
peptide bonds on protein backbone
rigid peptide bond = C-N
rotatable phi = N-C alpha
rotatable psi = C alpha - C
can change config by rotating phi or psi (some more fav than others)
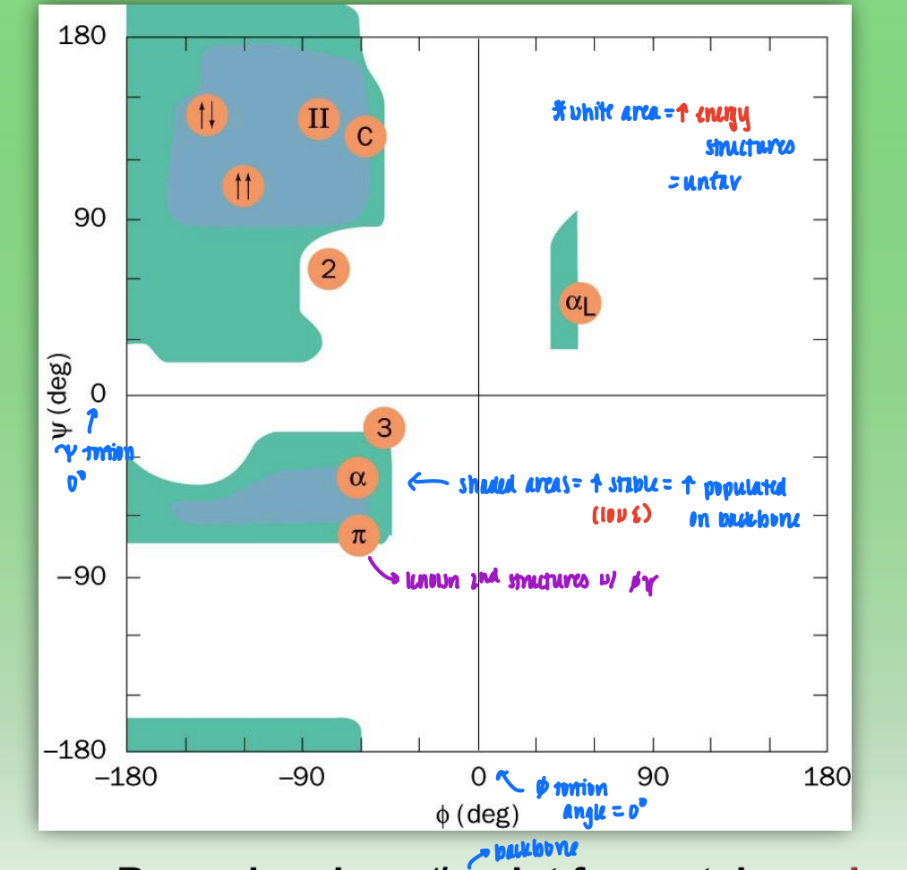
Ramachandran phi/psi plot for proteins
shaded areas = very stable (low E) bond configs = highly populated in backbone
white area = high energy, unfav configs
not every combo fav = sterically inhibited
axis = tortion angles
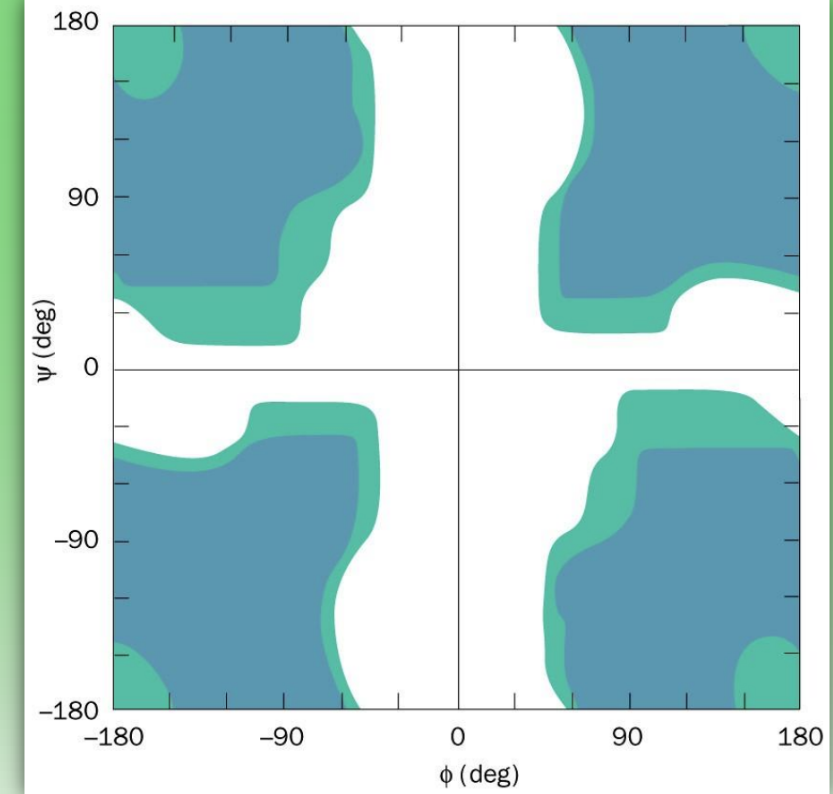
Ramachandran phi/psi plot for Gly
more shaded areas than normal = many more fav conformations
only have H side chain = no chiral center = less steric hindrance
= more flexibility to amide and can adopt phi/psi configs that other amino acids cannot
= high mobility & highly disordered 2nd struc
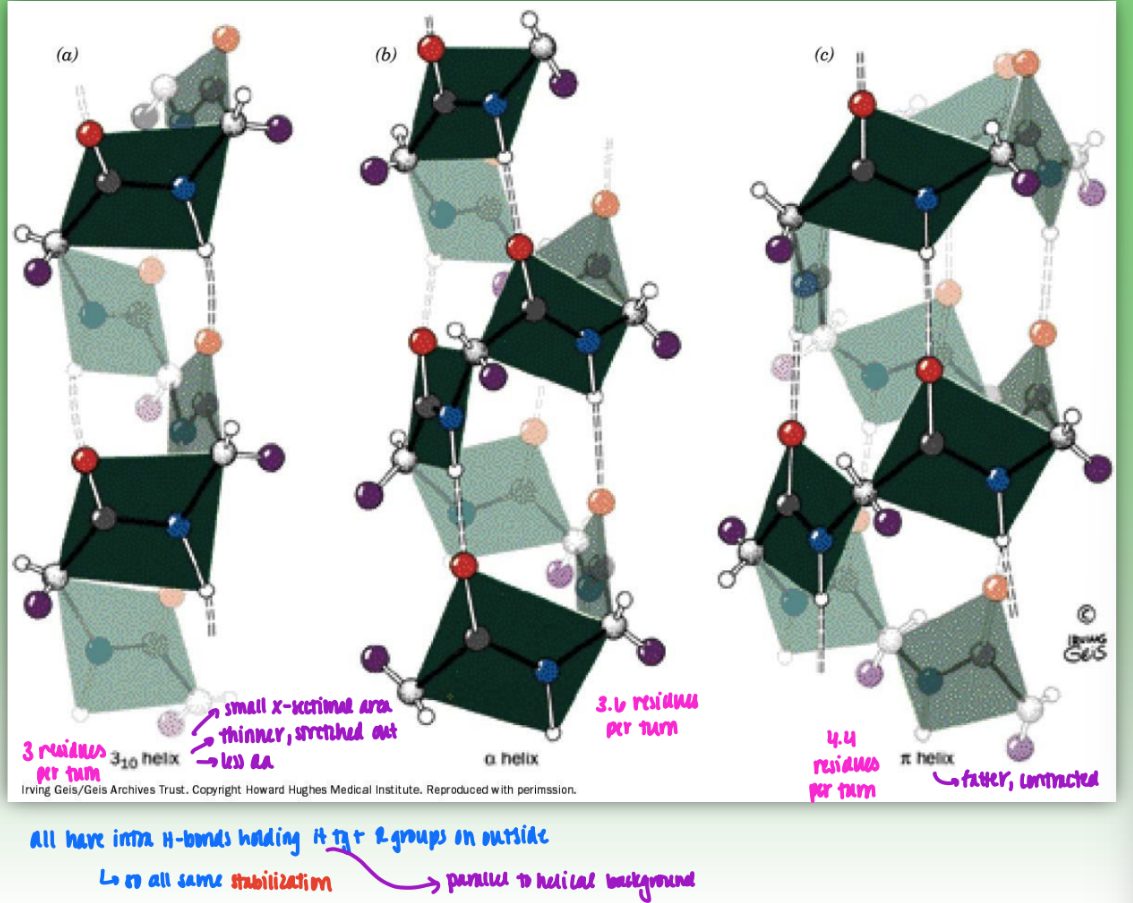
alpha helices
H bonds w/ carbonyl & N
R groups pointing outward = easily accessible
due to local folding
residues = big number = # of residues per 360 rotation
rise = overall rise
small number = # of atoms in H-bonded ring (how many atoms sep O from H)
comparing 310 , 3.613 (alpha) , 4.416 (pi) helices
310 = 3 residues per turn
small cross-sectional area
thinner, stretched out
less amino acids
3.613 = 3.6 residues per turn
4.416 = fatter, contracted
all have intra H-bonds holding H tg & R groups on outisde
so all same stabilization
analysis of alpha helix R-group composition
extract piece of backbone of myoglobin
determine location of e-helix = R group analysis
prot recog R group orientation (on outside surface of helix)
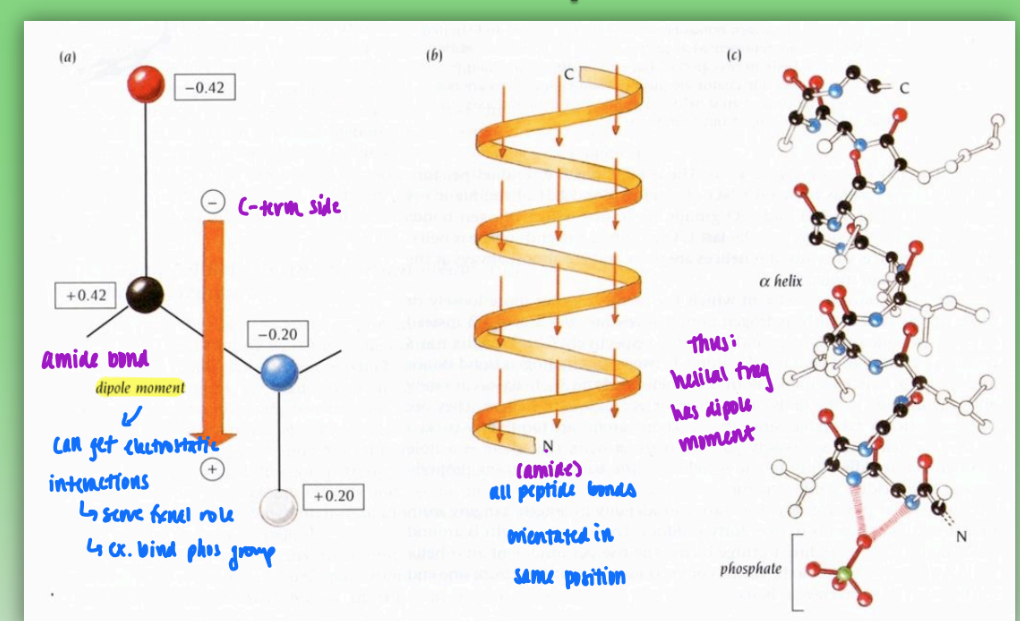
dipole moments in helices
fractional charges between neg c-term and pos n-term
all peptide bonds orientated in same positions = dipole moment in helix
can get electrostatic interactions
lead to fxnal role
ex. phosphate group binding
beta (pleated) sheet
primary struc interacts to fold in on itself to align like beta sheet
H bonds stabilize NH and carbonyl O
R groups on top or bottom of sheet surface → determines fxnal properties
can make amphipathic by using differntly charged R-groups on each side
antiparallel beta sheet
more stable = intra H-bonds more strongly aligned
each carbonyl can be acceptor for 2-3 bonds
one sheet N→C and one C←N
parallel beta sheet
both in same orientation C←N
intra H-bonds offset (not as perfectly aligned) = less stable
non linear H-bond = weaker
beta sheet stacking
most rigid structure = Ala & Gly
very strong
beta can easily stack (intercalations) (conformations of R groups highly complementary)
H and methyl pack tightly
types of beta sheet connections
hairpin = antiparallel (just need single turn)
stabilized by 1 H-bond
2 aa in turn but use C alpha 1 and C alpha 2
alternate turn by rotating tortion angles
out of plane crossovers = above & below plane (parallel)
more work / more complicated turn
gamma turns
caused by proline = ring struc induces bend
limits geometry & introduces bend / kink
C alpha 1 and C alpha 3 involved
omega loop
backbone bending motif
out of plane crossover of beta sheet
fibrous proteins
insoluble
usually comprised of 1 type of secondary struc
types: alpha keratins & collagens
alpha keratin
start as dimer
n-terminal head
coiled coil rod = alpha helix wrapped tg
c-term tail
connected via hydrophobic interface (dissymmetric distribution of aa)
dimer interface due to hydrophob primary struc
protofilament
head and tail more globular = can self associate to form protofilament
microfibril = disulfide bonds lead to formation and stablization
collagen
very strong fiber = used in connective tissue
distinctive amino acid comp: Gly, Pro, Hyp
Hyl also present (involved in cross-linking & glycosylation w/ O)
triple helical structure = 3 polypep chains wrapped around one another
stabilized by H-bond at interface
Gly = contrib NH (donor)
Pro = contrib O (acceptor)
glycosylated at Hyl residues w/ Glc-Gal post-translational
covalently cross-links Lys, Hyl, and His side chains in collagen
x-ray diffraction
image produced from x-ray scatters
uses crystal of myoglobin (static state)
this image = well-behaved = each prot molec in crystal is identical = good resolution
compare multiple diffraction patterns = create 3D structure
lower resolution = hard to determine structure
h-bonding = no diffraction so must rely on heavy atoms
must have good resolution so can differentiate between atoms
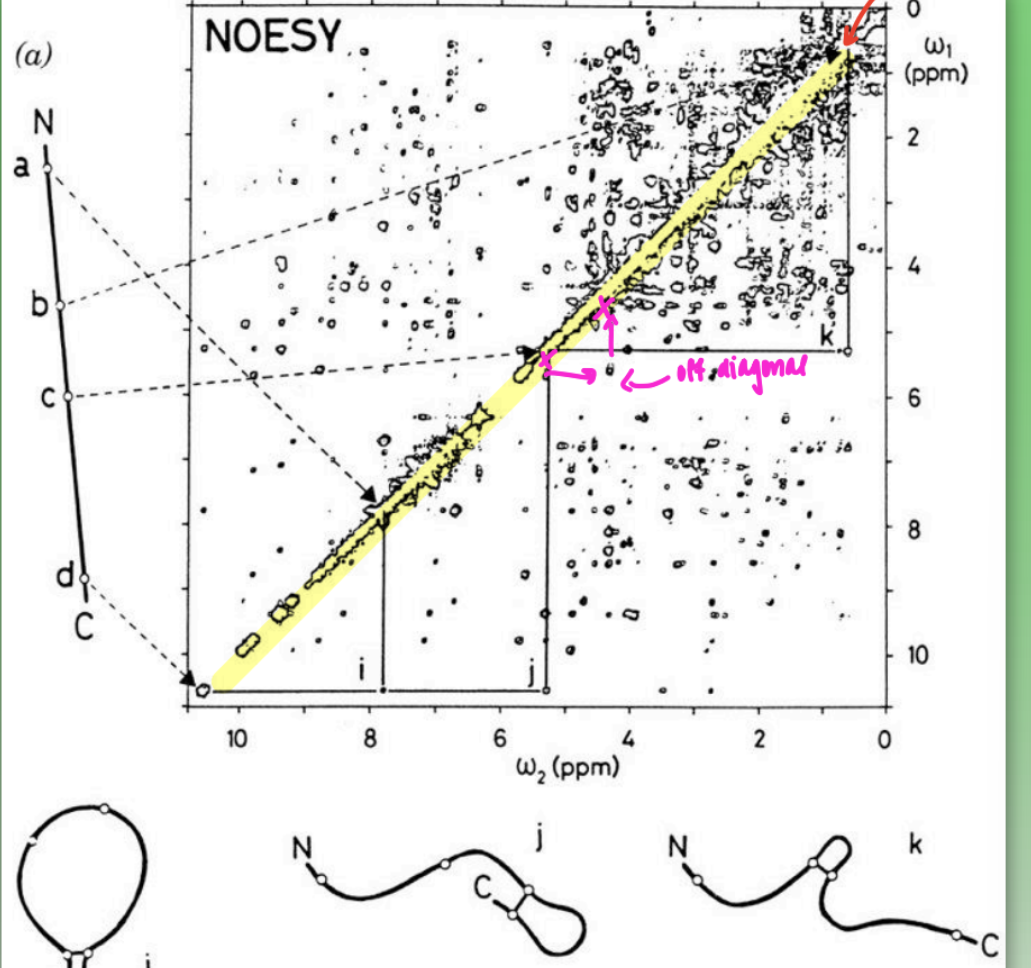
2D NOESY spectra
must use 2D spectrometry to determine protein internuclear distances
1D = too many overlapping signals
use “off-diagonal” method to determine internuc dist of all protons ( r )
many distances = put into computer program = determines how prot must be folded to account for all internuc distances
shows natural aq state behaviors
inconsistent fitting (less overlap) = intrinsic disorder in dynamic prot or insufficient number of distances
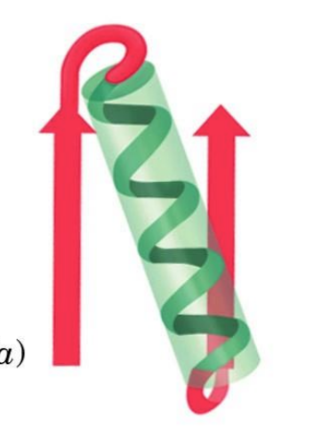
helical wheel
represents distribution of polar & nonpolar residues in helix
helix = amphiphilic = one side np & one side polar
inner facing proteins = hydrophob = hide from aq environment
asymmetric primary struc of aa leads to folding of 2/3 structure
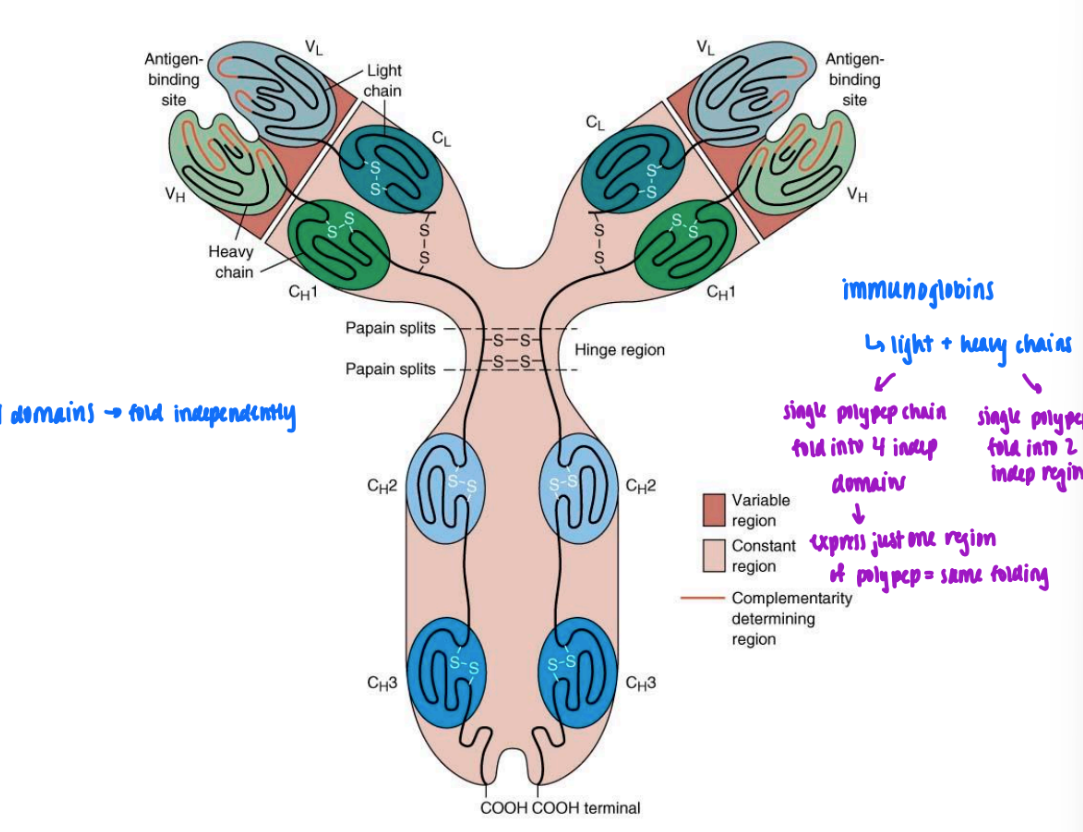
domain definition
long peptide chain can fold into single peptide domain (like myoglobin)
or single polypep chain can fold into 2 domains independent of one another
will fold same way or w/o presence of other = folding solely based on primary struc
if can truncate polypep & one express one & still folds same = domain
ex. immunoglobin
light & heavy chains
single polypep chains that fold into 4/2 indep domains
beta - alpha - beta
super secondary structure
parallel beta sheet
uses helix as cross over
stabilized by H bonds
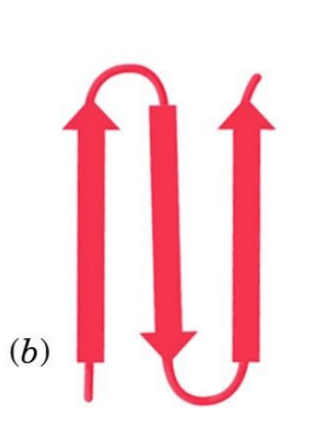
beta - hairpin
super secondary structure
2 anti parallel beta
fold in on each other to produce hairpin turns
stabilized by H bonds
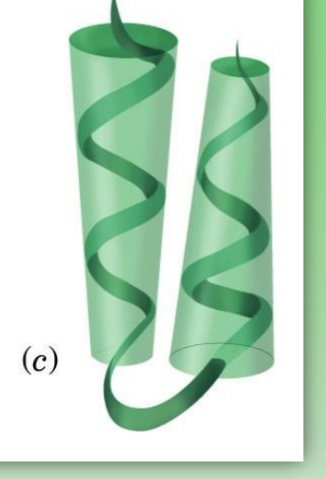
alpha - alpha
super secondary structure
alpha and alpha separated by turn
stabilized by dissymmetric distrib of hydrophob/phil interactions at interface
like alpha-keratin
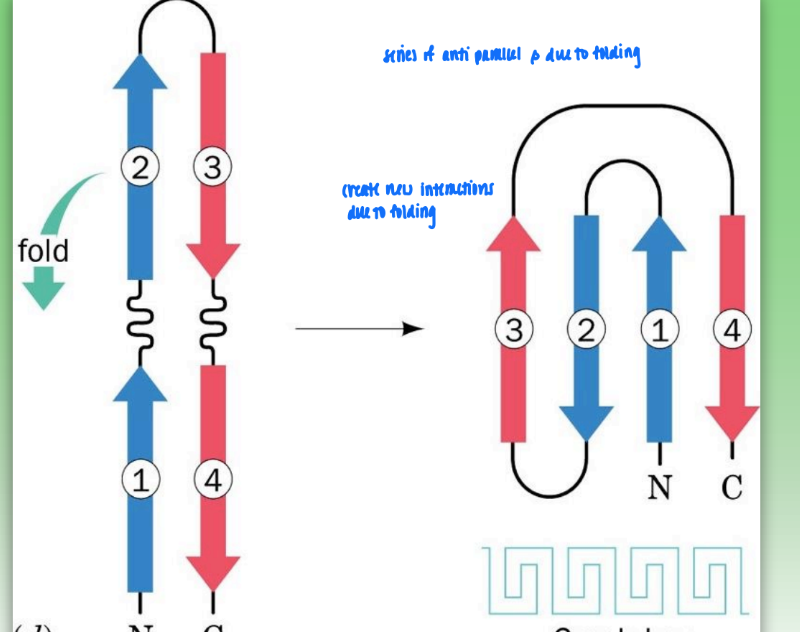
greek key motif
super secondary structure
series of antiparallel beta due to folding
create new interactions from folding
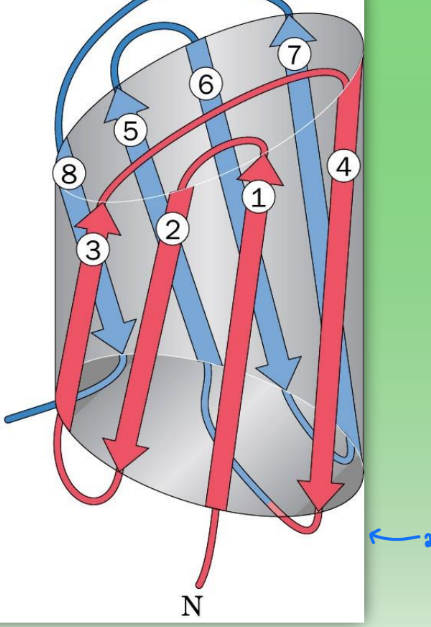
beta barrel
super secondary structure
2 greek key motifs
cylindrical → pairing introduces pore/hole
found in memb-bound proteins where hole is needed to allow things to pass thru memb (transport protein)
Cro dimer
dimer that binds DNA duplex
dimer = fxnal form
monomers cannot bind DNA duplex
H bonds hold each monomer tg
dimerization stabilized by antiparallel beta sheet interface
protruding R groups on alpha helix available to perfectly penetrate into major & minor groove of DNA
stabilized w/ H-bonding
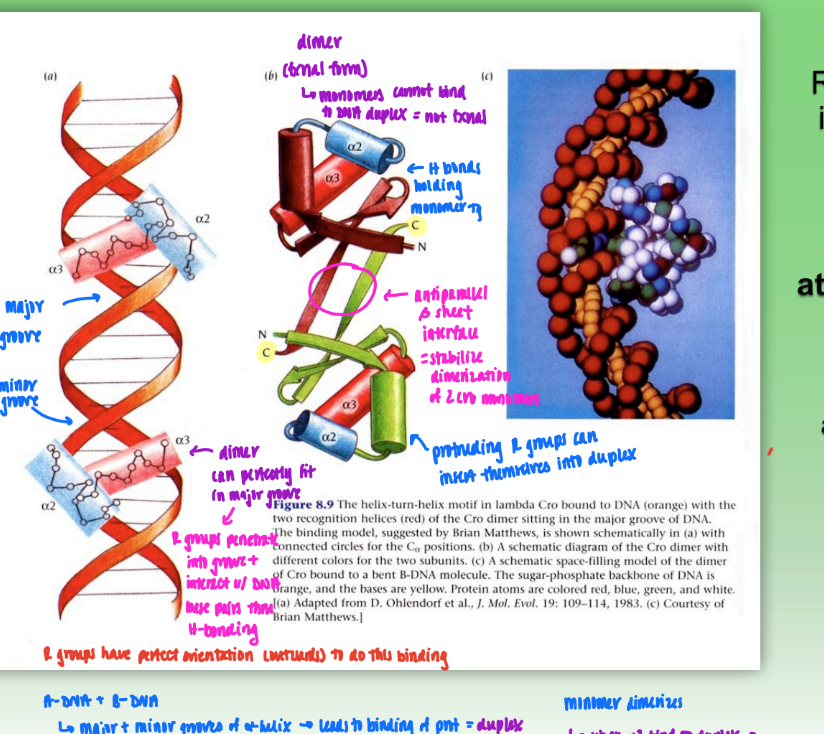
zinc finger protein
combination of beta sheet, alpha helix, and random coil
Cys-Cys-His-His family
2 His = imidazole ring
2 Cys = S
Zn anchors structures and forces fold
very strong ability to bind protein → often attached to DNA
when multiple zinc fingers bound tg = at least one able to bind to DNA
via alpha helix and protruding R groups w/ specificity to bind grooves
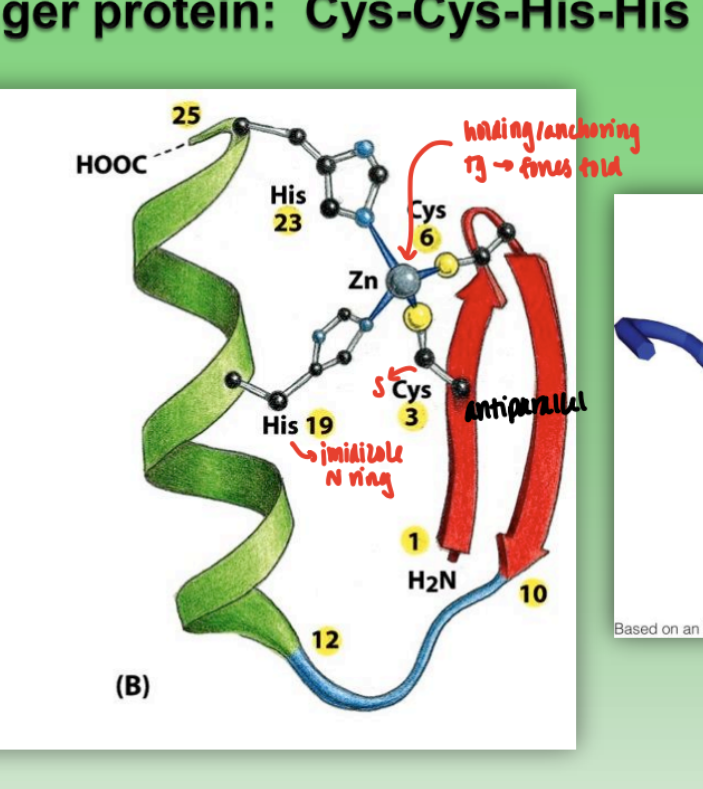
leucine zipper
dimer bound to DNA
2 alpha helix bound tg by Leu-Leu interface
not hydrophob residues!!!
protruding R-groups bind to DNA
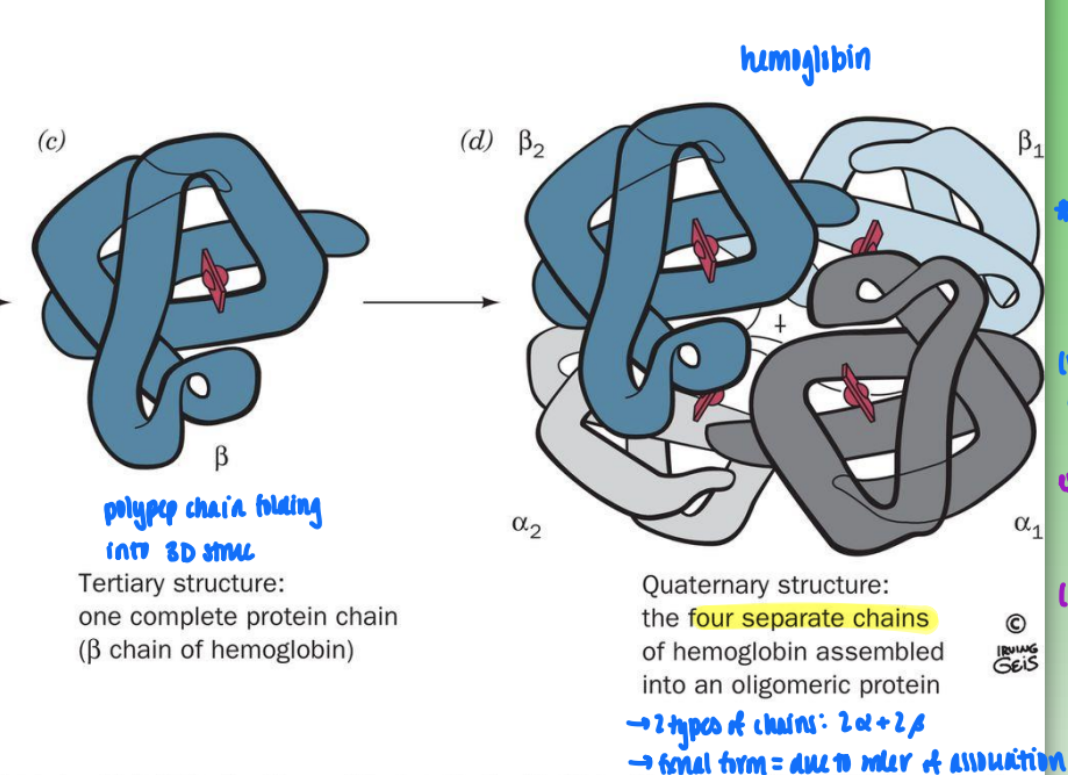
structural hierarchy in proteins
primary (amino seq in polypep chain)
secondary structure (helix)
tertiary structure (polypep chain folding into 3D struc)
quaternary structure (four sep chains associating thru non-cov interactions
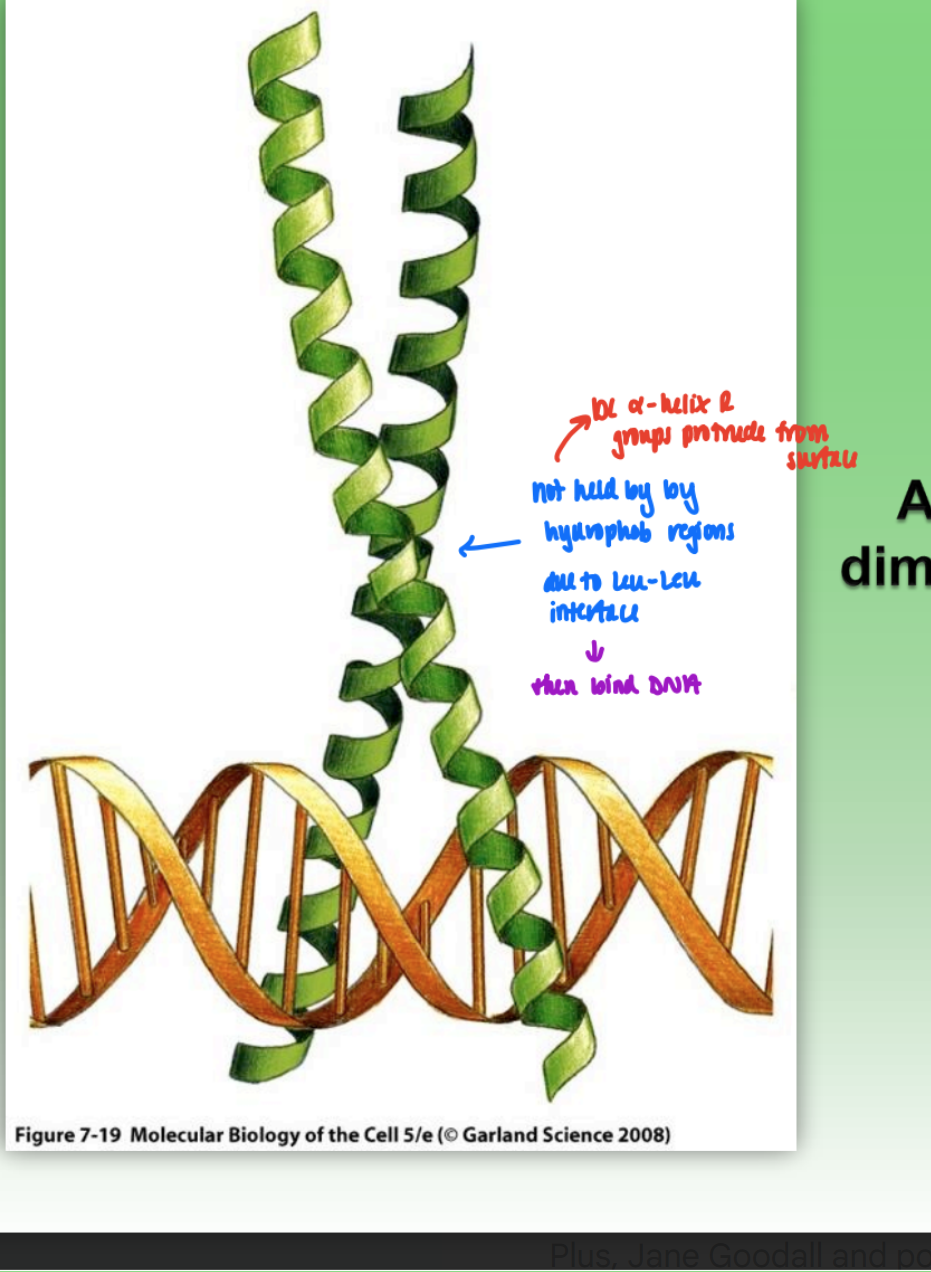
quaternary structure (hemoglobin)
2 types of chains: 2 alpha & 2 beta
fxnal form = due to order of association
associated thru noncov interactions (very weak so easily disrupted)
hemoglobin = O2 binding prot
binding to one site in one domain effects entire molec
binding = leads to struc change = changes affinity of all other sites
= cooperativity (communication)
differs from single domain in 3 struc (myoglobin) = independent binding (O2 binding one sit does not effect others)
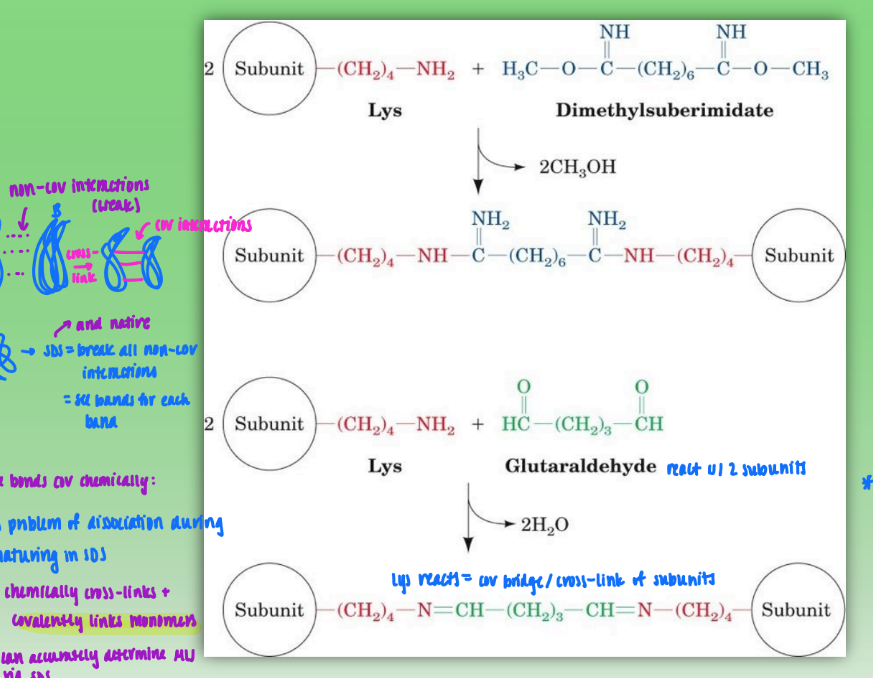
chemical cross-linking of oligomeric proteins
start w/ 2 subunits of quat prot & dimethylsuberimidate or glutaraldehyde
Lys on subunits reacts & creates cov bridge / cross-link between subunits/monomers
allows quat prot to undergo SDS or native so can properly determine molecular weight of proteins
SDS denatures and breaks down non-cov bonds that normally hold quat struct tg
get multiple bands (one per secondary struc)
limitations: concentration important
too much = cross linking of other dimers = create new, unwanted tetramers
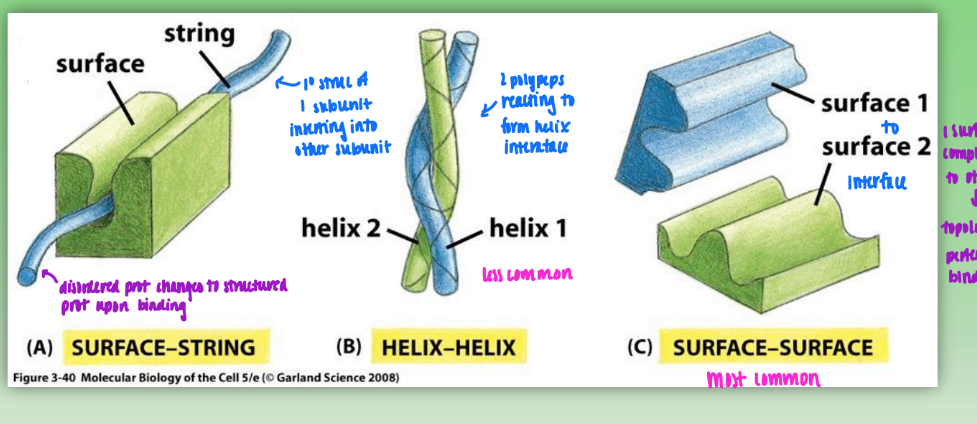
protein-protein interactions
surface-string
primary struc of 1 subunit inserts self into another subunit
helix-helix
2 polypeps react to form helical interface
surface-surface
1 surface complementary to other = topology allows perfect fit/binding of interfaces
most common
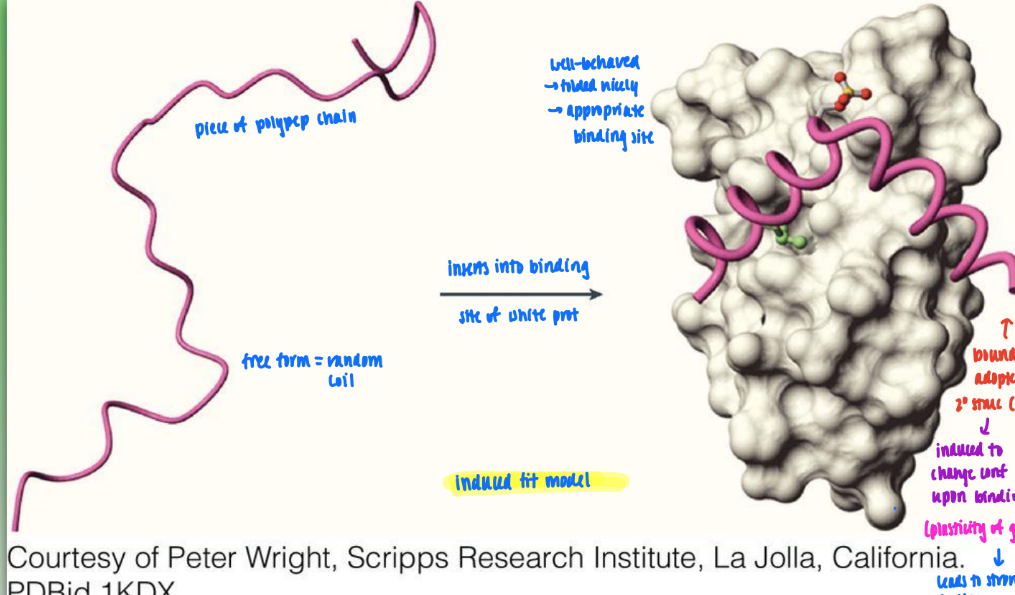
induced fit model
free form (random coil) induced to change conf to adopt defined secondary struc upon binding
shows plasticity of geometry
leads to stronger binding upon fold
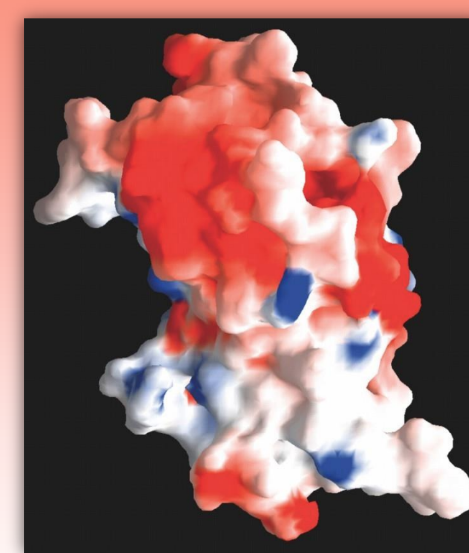
GRASP
graphical representation and analysis of surface properties
uses colors to determine distributions of ionic charge
ex. red = neg, blue = pos, white = neutral
can use to predict how prot will interact w/ other charged molecs
computer-derived molec
Chou-Fasman method
set of rules that allows you to determine what secondary structure will arise based on characteristics of primary structure/amino acid composition
uses frequency and propensity
input primary struc into computer = determine where alpha, beta, and turns will occur based on polypep chain & propensity & hydropathy values
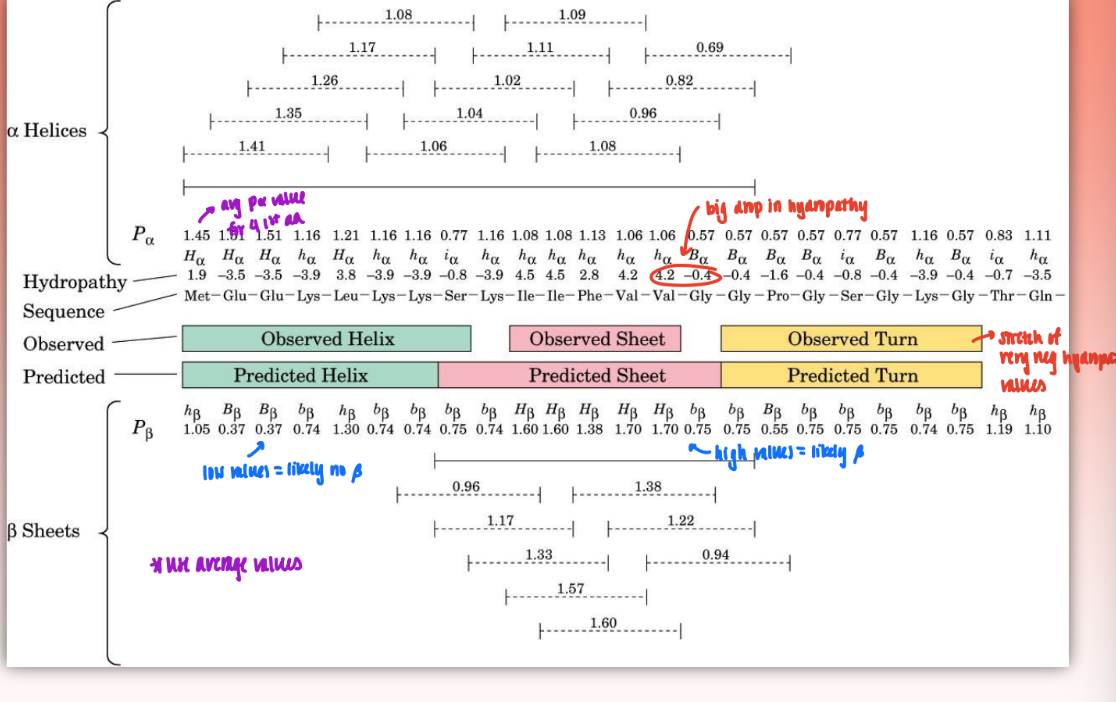
frequency
the statistically derived likelihood or propensity that a specific amino acid will be found in a particular type of protein secondary structure
greater frequency = greater likelihood amino acid will contribute to forming structure
= f alpha = n alpha / n
n alpha = number of amin acid residues of the given type (ex. alanine) that occur in alpha helices
n = total number of residues in of this type (ex. total number of alanines in protein set)
propensity
the tendency of an amino acid to prefer one type of amino acid structure over another
determined by: P alpha = f alpha / <falpha>
<falpha> = average value of f alpha for all 20 residues
when P alpha > 1 = residue occurs w/ greater than average frequency in an alpha helix
1 = average frequency
if p very low = strong break = usually Proline or Glycine (induces kink & disrupts secondary structure)
changes over time as more prot analyzed = living study
Rose method
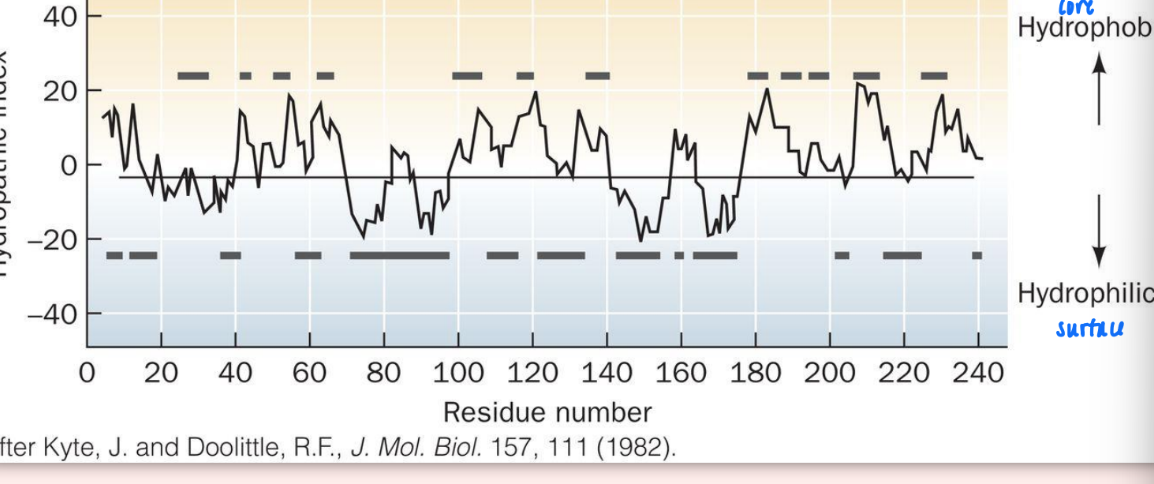
occur on surface of protein
occur at positions along a polypep chain when hydropathy goes from max to min (excluding helical regions)
predicts where turns in struc will occur
hydropathy scale
very high positive values = very hydrophobic (interior)
very negative = very hydrophilic (exterior)
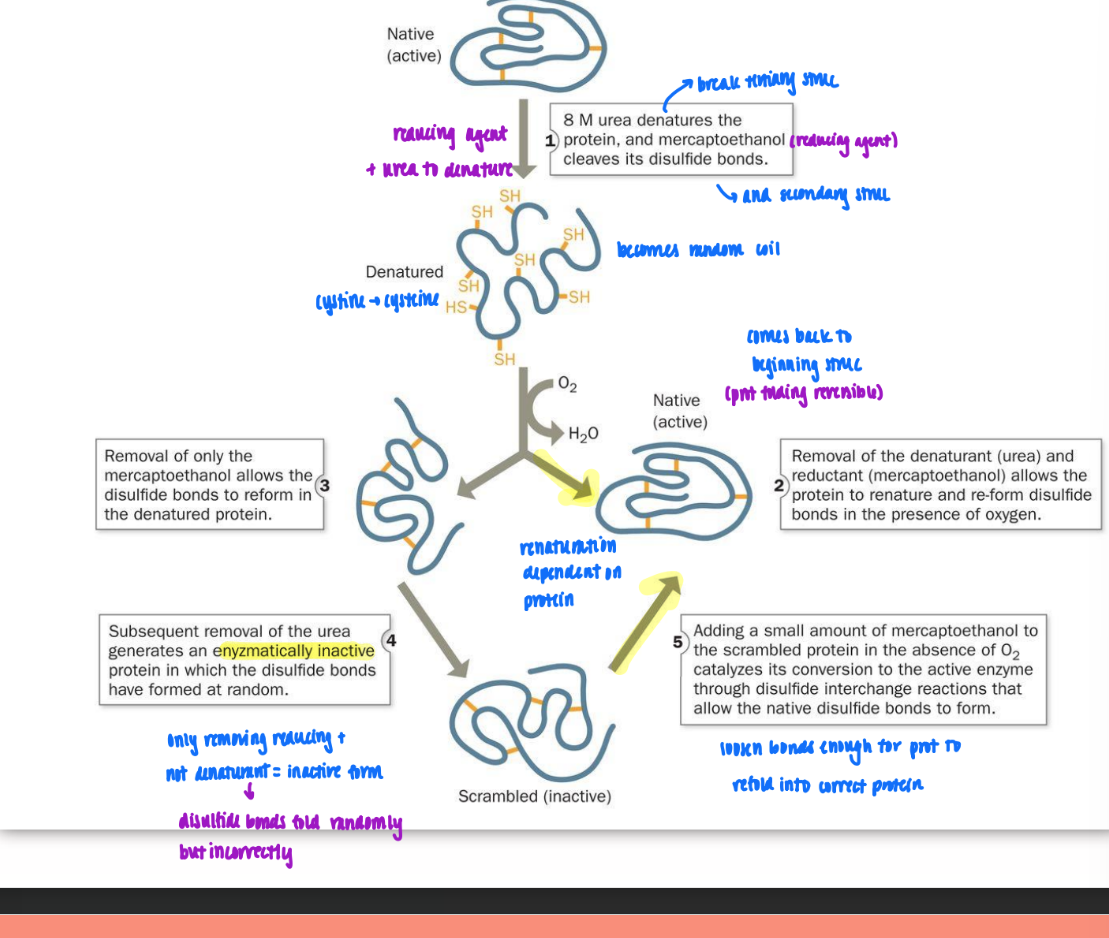
reductive denaturation & oxidative renaturation
use reducing agent (mercaptoethanol) & denaturing agent (urea) to break down tertiary and secondary struc = become random coil
reduction = remove disulfide bonds (S-S to SH)
denature = unravel (cystine → cysteine)
remove both at once = back to same (FOR RNase and some prot)
remove just reducing agent = disulfide bonds form in wrong places
then remove urea = enzymatically inactive prot generated
have to add small amt of reducing back to loosen bond just enough for port to hopefully refold into correct protein
heat-induced protein denaturation curve (for RNase A)
plot fractional change against Tm
fractional change = any measured parameter
S-shaped curve = implies cooperativity
once begins denaturing at one site = everything begins to denature
steeper slope = greater cooperativity
denaturing & renaturing = reversible process
due to temp (denature w/ heat & renature upon cooling)
conclusion: everything polypep needs to fold is in primary struc
catalytically active after each transition thru curve
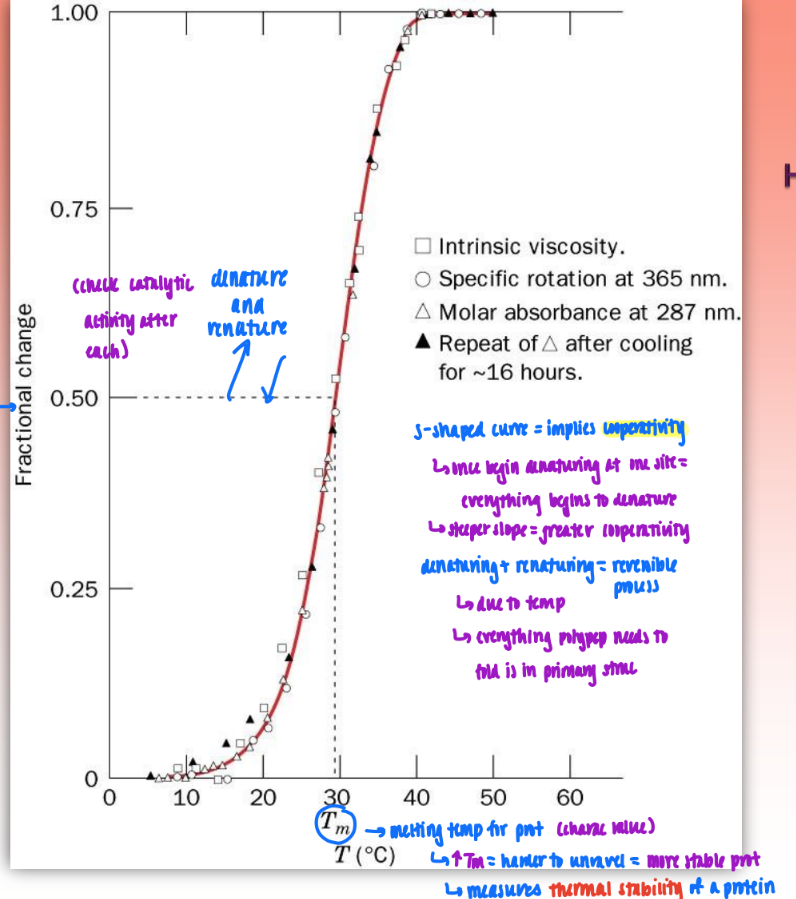
Tm
melting temp for proteins
characteristic value
measures thermal stability of protein
higher Tm = harder to unravel = more stable prot
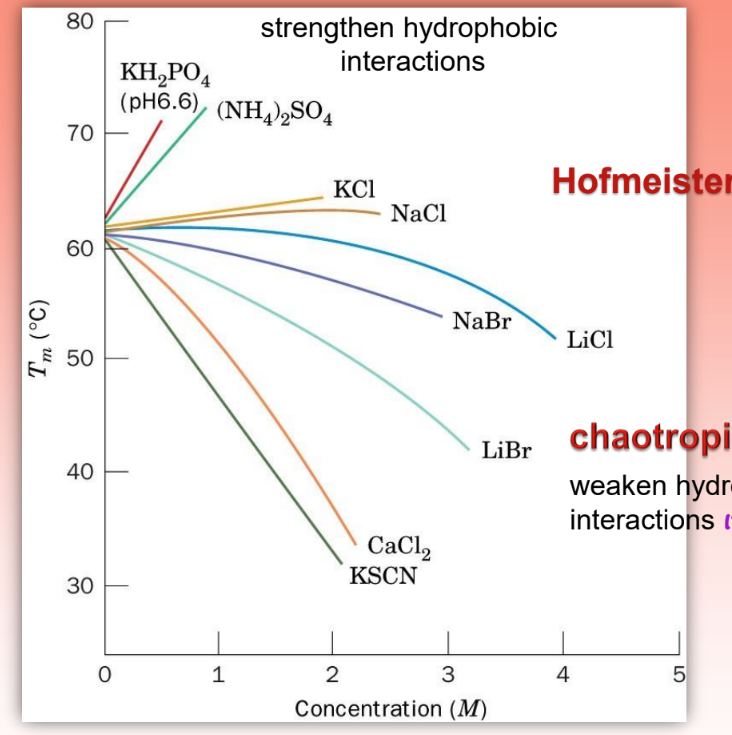
Hofmeister series
shows the effect of concentration and slats on Tm
chaotropic salts = reduce Tm & destabilize protein
make easier to denature & unravel prot by weakening hydrophob bonds
some determinants of protein folding
helices/sheets predominate in proteins bc they fill space efficiently
why prot density = same
secondary struc pack very efficiently
prot folding is directed mainly by interna; residues (protein folding is driven by hydrophobic forces → the hydrophobic effect)
hydrophob must be folded in core
immobile H2O molecs in unfolded prot in core → more mobile in aq
increases S = neg G
prot folding dep on primary struc (amino acid sequence)
but not case for all prot → may have other influencing factors
slow rate of folding = post-trans folding
fast rate of folding = folding during trans (on ribosome)
hydrophobic effect & protein folding
ex. fav folding for hydrophob molec moving from water to hydrophob solvent
H = post
S = neg
water molec release from hydrophob molec = increase entropy
= -G
fav to fold (denature prot to folded prot)
Levinthal Paradox
crude estimate of the time required for protein folding
conclusion: proteins must fold via ordered pathway or set of pathways bc time for random folding = greater than age of universe
based on conformations of phi and psi (not based on Ramachandran bc unfold prot)
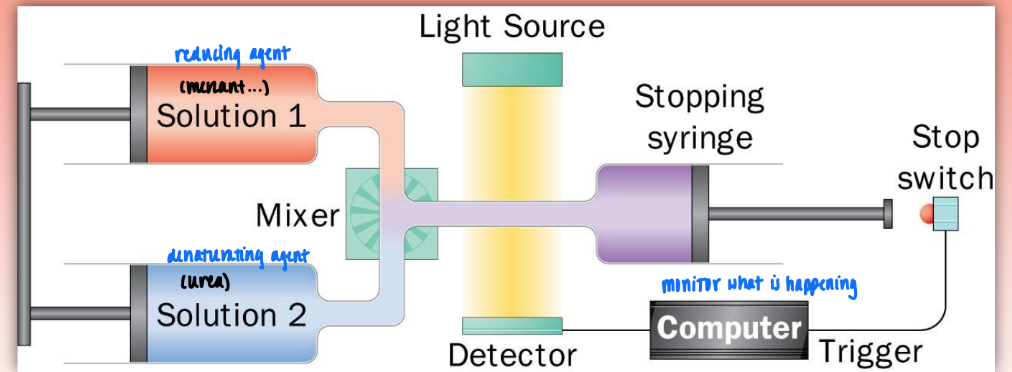
stopped-flow device
very fast way to mix two solutions (very low dead time)
ex. reducing & dentauring agent
then observe prot folding in detector
denat/renat or earlu stages of prot folding
vary dead times to observe dif stages of prot folding
detection/analysis via:
UV absob (look at aromatic aa = primary struc)
CD spectrum (measure secondary struc) = better method