molecular genetics unit 3/final study guide
1/139
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
140 Terms
name a primary mechanism of gene regulation
regulating the ability of RNA pol II to bind to the promoter — increased binding = increased transcription and therefore increased expression; i.e. using GTFs (euks) or sigmas (proks)
tell the transcription initiation story for proks
RNA pol in proks can bind decently in seq-non-specific manner. part-time sigma subunits bind upstream of the promoter to increase binding stability of RNA pol at the promoters by creating more contacts for it. there are many different sigmas and promoter patterns, but the one responsible for driving the most interactions is sigma 70
there are 2 sequences responsible for sigma 70 binding, which are physically separate from each other. one is at position -10 (10 bp upstream of the transcriptional start site) and position -35. these sites exhibit significant amount of similarity, enough to be recognized by sigma 70

describe each of these possibilities for a sigma 70 promoter in e coli
A: standard promoter for sigma 70 in e coli
B: some promoters have an upstream element that is productive for sigma 70 binding
C: some promoters may have an extra-long -10 binding site in place of having the -35 site
D: some will have discriminator sites that make it so that either sigma 70 can bind or so that it can’t bind (compare to LAC operon from genetics)
tell the transcriptional initiation story for euks
first: gonna be multiple binding sites, more than the 2 in proks. there are sequences upstream like proks, but ALSO downstream! thus for RNA pol II to bind, it is likely making functional and sequence specific interactions downstream, upstream, and at the initiation site simultaneously
thus just like in e coli, the RNA pol II is attracted to specific sites in the DNA. most famously, ~30 bases upstream we see the TATA box which binds to the first/most famous GTF of all: TATA binding protein, TBP
then, a complex of GTFs assemble with the RNA pol near the promoter. when TFIIH phosphorylates the pol tail, it can leave the complex and bind the DNA at the promoter
three ways that GTFs can act
some GTFs bind directly to DNA and recognize seq associated with promoters.
some bind to the proteins that bind to DNA and link them to the RNA pol II complex
others modify the RNA pol II complex in a way necessary for initiation of xcription
true/false: there are more contacts for proteins in promoters for prok genes compared to euks
false; there are far more protein contacts in euks
how should we think about GTFs’ roles in gene expression in euks?
for our purposes, ALL of the GTFs are REQUIRED for expression of EVERY protein-encoding gene
not the full story though — missing the impact of chromatin structure, and the possibility that other proteins will bind to the genome and regulate the rate at which these complexes form — in proks AND euks!
tell the story of GTFs and the impact on transcriptional initiation in euks
TATA binding proteins (TBP) and TFIID sit down as a complex upstream of the transcriptional start. this flags the promoter for other GTFs and eventually RNA pol II
first they call in TFIIB and TFIIA— B binds to DNA directly in a sequence specific way; A does not, as its purpose is to be a scaffolding protein for RNA pol II to interact with the other GTFs.
RNA pol II is pre-bound to TFIIF, and together they sit down on the complex. TFIIF calls in TFIIE and TFIIH. H is of particular interest because it is a protein kinase, and will phosphorylate the polymerase complex’s tail, causing RNA pol II to let go of the rest of the complex and sit on the promoter in a sequence-specific way
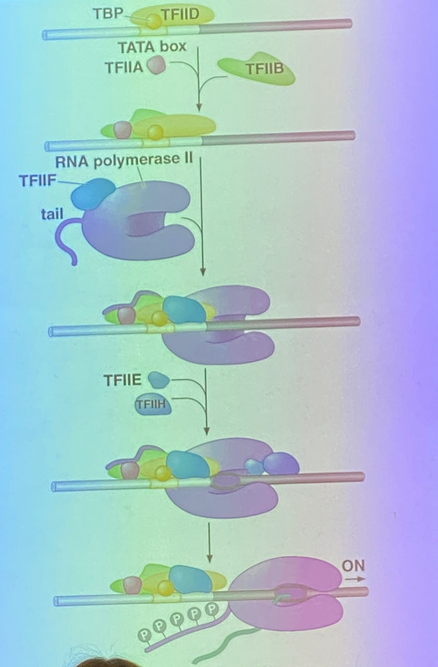
list the GTFs involved in eukaryotic transcriptional initiation and their purpose
TBP + D →→ B + A →→ pol + F →→ E + H
TBP and TFIID — sit first, call in B and A
TFIIB — binds DNA in seq specific way
TFIIA — scaffolding protein; necessary for RNA pol II to interact with the other GTFs
TFIIF — pre-bound to RNA pol; brings in E and H
TFIIE — comes in with H
TFIIH — phosphorylates the holoenzyme complex’s tail so RNA pol II can leave and bind instead to the promoter in sequence-specific way
e. coli lactase experiment: if no galactose, glucose only: expression of lactase is low. if we spike it with lactose, what happens? what does this suggest?
eventually see lactase expression only when glucose is depleted — only then do they turn on the cells necessary to digest lactose. suggests that it has the genetic circuitry to detect what sugars are present, and to prioritize glucose metabolism until it’s gone
e. coli lactase experiment: how can the bacteria detect the sugars present and prioritize one over another?
works at level of transcriptional initiation. there are proteins that are sensitive to the sugar levels, proteins which affect the rate of RNA pol binding to the promoter.
there are also transcriptional regulators, which may be necessary in some genes and not in others
discuss transcriptional regulators — define? in proks and/or euks, and how do they typically act?
proteins that enhance or inhibit gene expression, usually by affecting RNA pol’s binding ability. activators act positively, and repressors act negatively. they are NOT GTFs
in both proks AND euks
in proks, usually bind to sites very close to promoters (potentially even overlapping the promoters!)
in euks, they often bind very far away from the promoters — ties in to chromatin looping. TRs may require enormous loops to account for the great distances!
true/false: in euks, most activators interact directly with the RNA pol or the GTFs to enhance binding.
false — instead, they interact with the mediator complex, which is REQUIRED in all eukaryotic cells.
many other intermediate proteins, including the histones, act between the DNA-bound transcription regulator and RNA polymerase.
what is the relevance of the mediator complex?
TAs in proks bind directly to the polymerase, but TAs in euks rarely do. instead, they interact w the mediator complex, which interacts with the polymerase.
comprised of MED proteins (which may vary from one complex to the next, but likely not huge source of differences in expression)
RNA pol is already huge, especially with the mediator involved — for a promoter to be used, it must be accessible by the ENTIRE complex, so nucleosome organization is really important
gives us a clear mechanism for driving differential expression of genes not only at promoters, but at enhancers too!
transcriptional elongation in proks vs euks
proks: RNA pol adds nucleotides to 3’ ends using watson-crick base pairing
euks: more complicated … requires processing; nucleosomes pose unique challenges; chromatin structure is involved in determining when start sites are used and the rate at which it goes to completion; RNA pol can fall off during transcription
4 impacts of chromatin structure on transcriptional elongation
RNA pol sometimes interacts with nucleosomes, as we do sometimes see gene expression near the nucleosomes even if the loop ends have greater expression.
transcription does NOT disrupt nucleosomal structure — “affects” it to a certain degree, but does NOT require histone displacement — RNA pol can transcribe around the nucleosomes!
chromatin structure dictates when promoters are used and the rate at which transcription occurs
we don’t know the degree to which chromatin packing affects the rate of RNA pol falling off
true/false: the stop codon dictates transcriptional termination
FALSE — stop codons are for translation, not transcription — there are NO clear arrangements of nucleotides that trigger termination in proks and euks
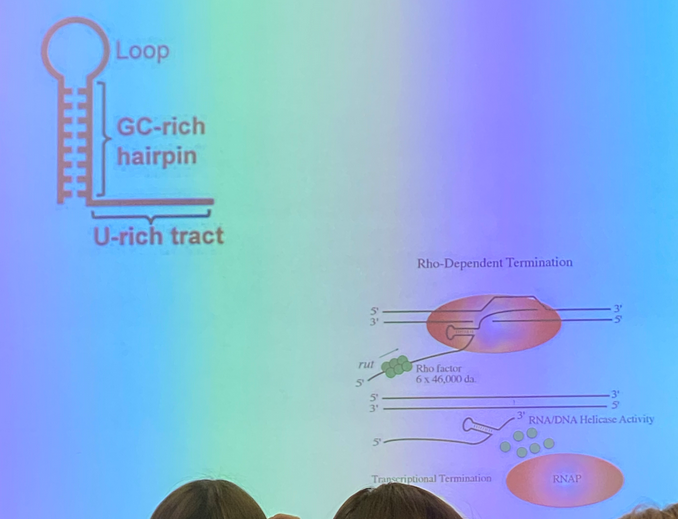
describe the two ways that we observe transcriptional termination in proks
rho-dependent: rho is a protein that binds in sequence specific way to “rut sites” in (some) e. coli scripts. rho proteins bind in complexes of 6. it moves from the rut binding site to a sequence motif that allows it to interact with RNA pol and make it fall off. trancription is thus terminated
rho-independent: form a 3D stem and loop RNA to form double-stranded character. this loop interacts with RNA pol, making it pause and lose processivity (decreases the likelihood of the next nucleotide being added and thus increases odds of the pol falling off)
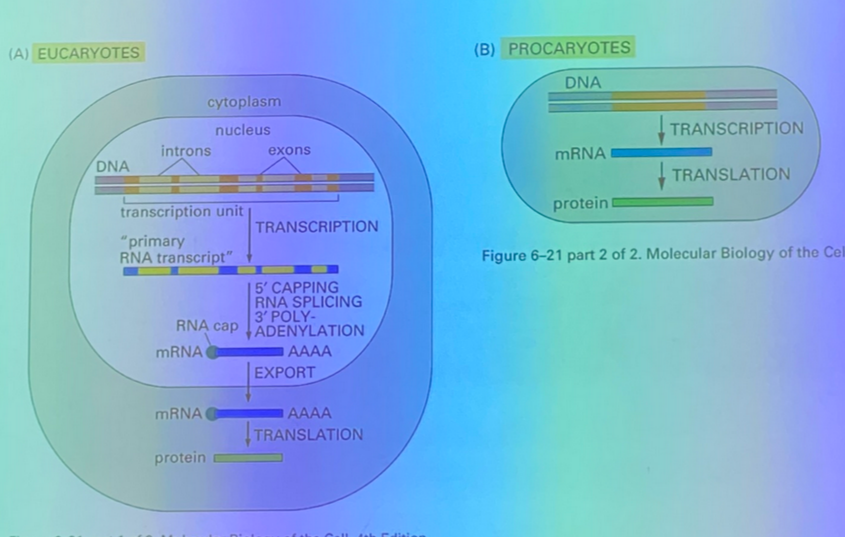
describe end processing of eukaryotic mRNA — what is the significance of the processed ends, and how is the poly-A tail formed?
there is a methylated G at the 5’ end, and the poly-A tail at the 3’ end. these two ends commonly interact with each other, so our transcripts are usually CIRCULARIZED
the poly-A tail comes from the polyadenylation A enzyme adding it on to all messages. an AAUAAA sequence is recognized by CPSF protein, which binds and calls in other accessory proteins (CFI + CFII) which bring in cleavage stimulating factor to cleave the script at a particular sequence as transcription occurs simultaneously downstream of the AAUUAAA. once it’s cleaved, polyadenylation A adds the poly-A tail, which is several hundred A residues long!
how does transcriptional termination occur in euks?
we really don’t know — two theories are that RNA pol just falls off somehow, or that cleavage affects the RNA pol biology to force it to fall off more readily
true/false: splicing occurs in proks and euks
false — euks only
why is splicing important? 4 reasons
allows us to make multiple proteins from one gene → higher degree of complexity; good to have multiple versions of proteins that are important to (early!) development
cell bio perspective: in humans (not all euks) it is REQUIRED for nuclear export (transport of mRNA out of nucleus). splicing creates a unique sequence at the junction between re-joined exons. the exon junctional complex (EJC) binds in a sequence specific way here, and hooks the mRNA up to the export machinery
necessary for complete processing of pre-mRNA
evolutionary perspective: give the chance for recombination events that wouldn’t be possible otherwise → diversity. if recombination in coding regions were random, there’d be a low likelihood that a functional protein would form.
true/false: introns can splice themselves out without help from a protein
half-true — some are self-splicing introns and essentially remove themselves from the genome, but don’t make up most of the introns in a complicated organism — none of the introns in humans are self-splicing, but the mechanism isn’t too different
which is longer on average, exons or introns?
introns — while exons average 100-200 bps, introns are more variable, with most being between 100-2k bps long in humans, but a few are greater than 30k long!
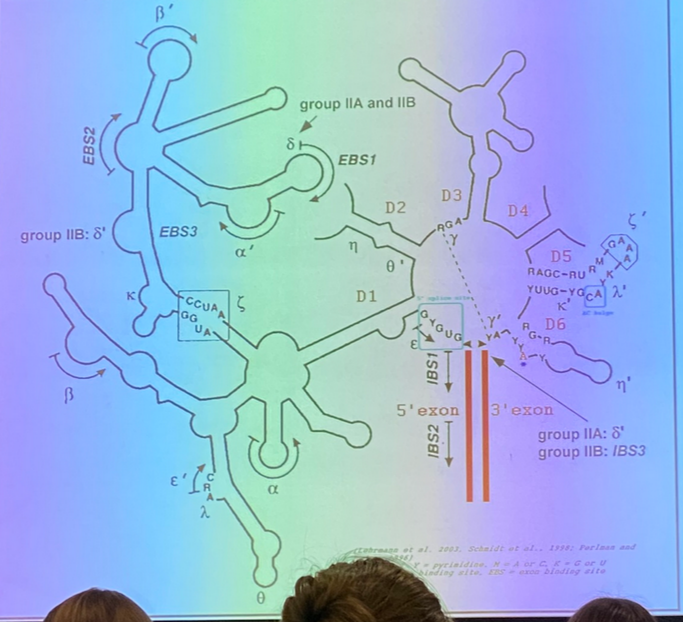
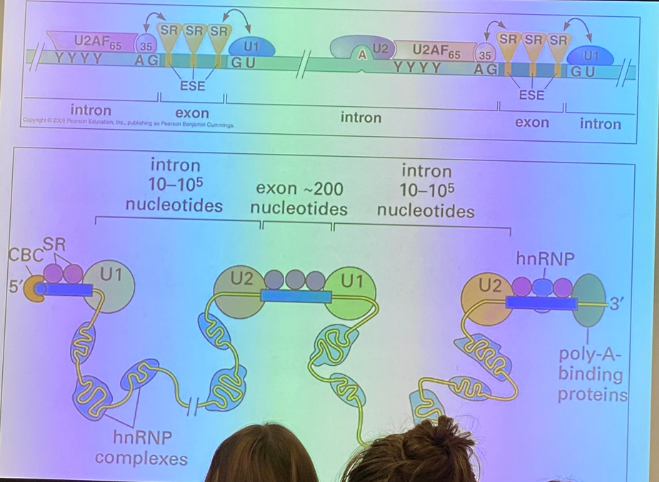
tell the splicing story — how do we identify introns?
there are stereotypical patterns at the junctions between it and the surrounding exons — the exon-intron (5’) junction, and the intron-exon (3’) junction.
we see classical sequences in both the exons and the introns themselves. in this image, Y11 is the consensus site, and indicates the presence of 11 pyrimidines in a row.
there is also the branch site, which is involved in the biochemistry responsible for removing the intron. it is found internally and spacing varies from one intron to the next, but has its own characteristics.

tell the splicing story — how are self-splicing introns removed, once recognized?
the branch site, the A residue, attacks the phosphodiester bond at the exon-intron junction, thanks to RNA’s ability to fold on itself. this junction becomes site-lysed, and we have interrupted the message by looping the intron, which will later be the lariat. the exon that got cut off now has a highly reactive OH group exposed, which attacks the intron-exon junction and kicks the intron off, now known as the lariat.
there is no enzyme involved here, but it’s enzyme-like… thus it is ribozyme-catalyzed, as it’s catalyzed by a molecule made of RNA rather than proteins.
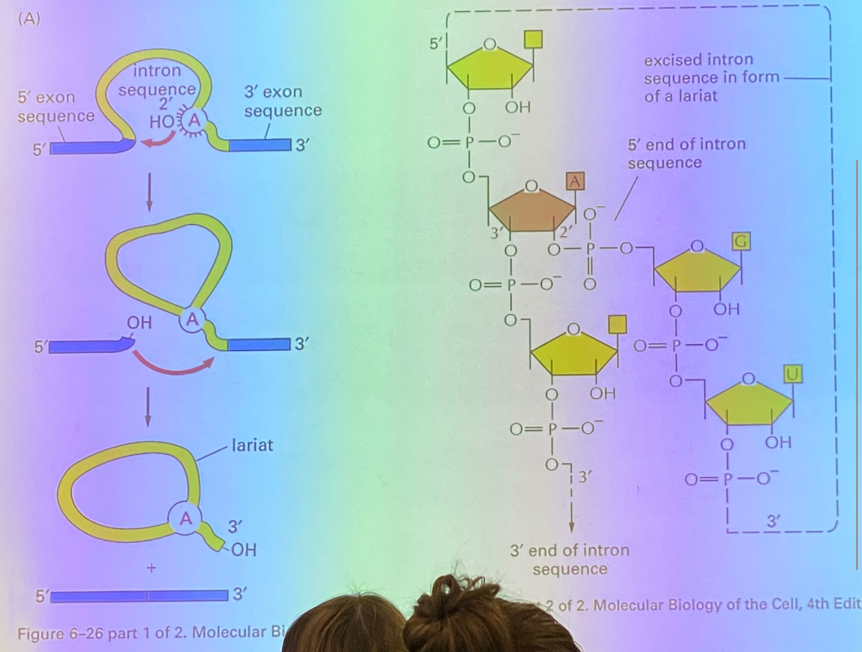
true/false: RNA can have discreet 3D structures associated with their activities
true. i.e. structural RNAs (rRNA, tRNA) resemble proteins and so they can behave like proteins
if it can catalyze a reaction like an enzyme would, it’s called a ribozyme
what is this image depicting?
a self-splicing intron
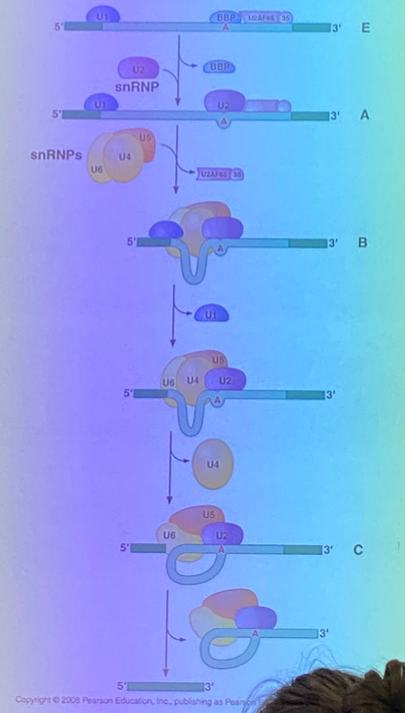
tell the splicing story — how are NON-self-splicing introns removed?
identification is the same — characteristic sequences at the junctions, and central A branch site.
require the spliceosome, which is a massive complex of accessory proteins+RNA molecules known as “small nuclear ribonucleoprotein” complexes, AKA snRNPS.
the snRNPS bring the splice sites close together so that the same mechanism seen in self-splicing can occur. the snRNPs bring the branch site to the 5’ junction, get a cyclized A residue, the exposed OH of the exon attacks the other junction, and the lariat is formed.
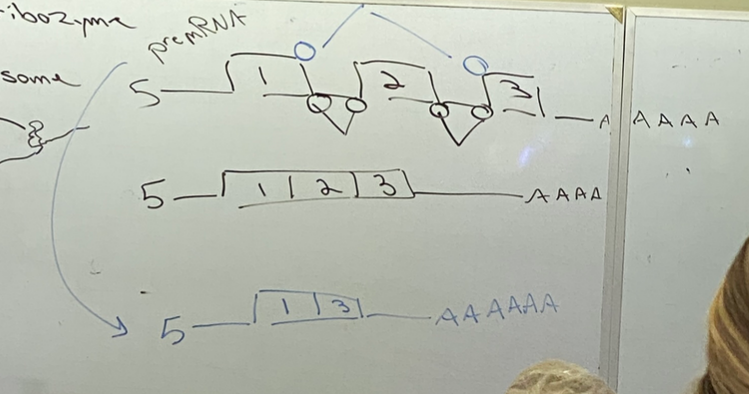
how do snRNPS open us up to the possibility of regulation?
can splice in many different ways, including by cutting out intervening exons between introns. different splicing leads to creation of a protein that is different, but likely has similar functionality to the one produced by the original transcript. not all combos work, so it’s not a random selection.
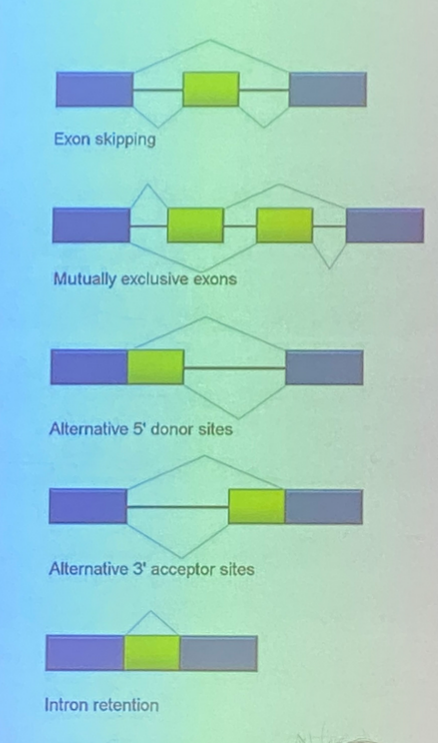
describe the different splicing events shown
1. can cut out just the introns, or chop out the intervening exon too
2. use one intervening exon or the other, but not both
3-5. the sequence in yellow can be thought of as either an intron or an exon, as it can be cut out or left in. it is an exon in name, as it can be retained, but it can also be cut out like an intron.
(5 looks confusing, but it’s essentially the same as the previous ones — there just isn’t a surrounding intron sequence that is guaranteed removal)
true/false: we can have proteins that direct snRNPs to/away from certain sequences so that they are or are not spliced in particular ways
true — SR proteins bind to mRNA exons and regulate the assembly of the spliceosome
as a whole, they build the spliceosomes. bind to message in context dependent way — regulated. it can do it differently in different cells, thus it regulates alt splicing.
what is the main difference between us and other animals, like wildebeests?
not the genome, but the order in which genes are expressed in early development — at least in part due to differential splicing events leading to different versions of proteins being expressed in different cells, or even in different regions of the same cell
true/false — neurons are not affected by differential splicing events
false — on the pre- and post-synaptic neurons, there are proteins that interact with one another to allow for synaptic transmission between the neurons. these proteins are subject to differential splicing events, and will vary from region to region or cell to cell. this affects how our nervous system works!
exons correspond more generally to protein domains — explain the significance of this
protein structure “rhymes” and there are very few structural domains seen despite all the diversity of life, i.e. ATPase domains, kinase domains, etc look similar from organism to organism.
for example, we see that the proteins involved in chromatin structure in euks are all made up of the same basic domains.
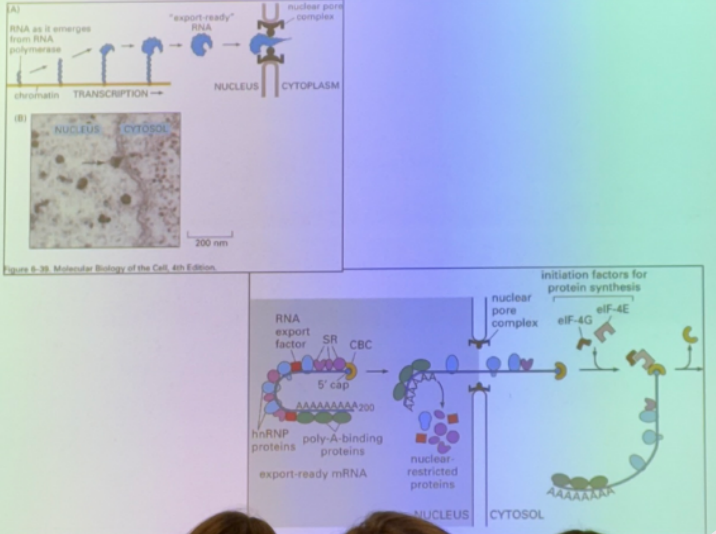
what is this image depicting?
the relevance of splicing for nuclear export
messages are not exported from nucleoplasm until they are spliced — sent thru nuclear pores only when processed. has a unique sequence seen only after splicing — at the junction between exons. proteins (EJC — exon junctional complex) bind in seq specific way at the exon junctions. hooks message up to machinery necessary for export.
lac operon: result when grown on glycerol and in absence of lactose, then spiked with lactose vs result when grown on glucose without lactose, then spiked with lactose
glycerol: in absence of lactose, do not make active b-gal. if spiked, it’s turned on immediately
glucose: in absence of lactose, no active b-gal made. when spiked, still no expression until glucose is depleted
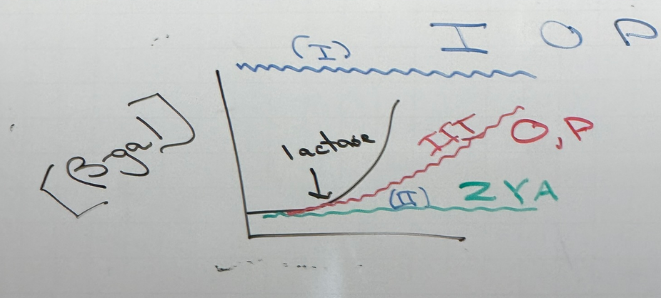
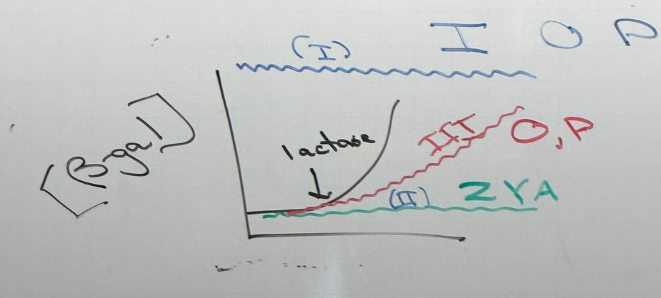
random mutations in LAC operon — what does each color show?
blue: class I mutations. cells always produce b-gal. these mutations land in I, O, P.
green: class II mutations. no produce of b-gal regardless of lactose concentration. land in Z, Y, A
red: class III mutations. b-gal production is delayed. found in O and P. simplest example: P is mutated so that RNA pol ii binds too strongly and can’t actually escape the promoter to initiate transcription, so transcriptional rates are lower/slower → delayed effect
note that these aren’t all necessarily mutually exclusive — there can be mutations in O/P that look like a class II. there can be the super-I mutation that has the same effect as class II. etc
do mutations in lacO and lacP result in overexpression, underexpression, both, or neither?
both
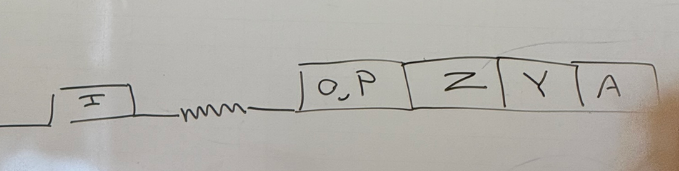
somewhere in this map of lac, there must be a gene to express b-gal, and somewhere there must be sequences responsible for the regulation (+ and -) of that gene. they couldn’t target each to mutate individually bc the tech didn’t exist yet, so what kind of test could they use? what’s the difference between cis/trans?
partial diploid analysis, AKA cis/trans test — see if any of them can act from far away, such as from an entirely different DNA molecule
any gene that can exert its effects from a different chromosome = trans = likely codes for regulatory protein; if it needs to be on the same chromosome = cis.
describe how a partial diploid analysis works
add a gene to e. coli using plasmids, and put together different combos of the wild vs mutant versions of the gene they had — I O P Z Y A.
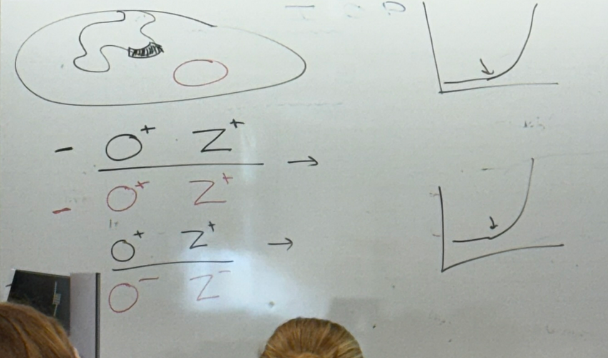
lac partial diploid analysis:
bacterial O+Z+; plasmid O+Z+?
bacterial O+Z+, plasmid O-Z-?
normal phenotype for both. appears as though wild type behaves in dominant way in the diploids
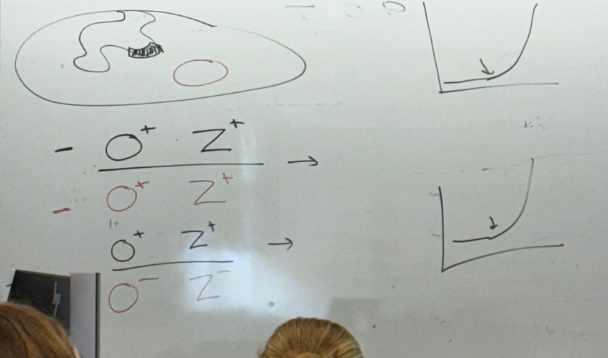
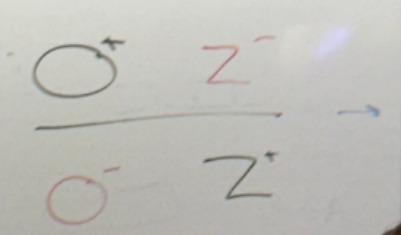
lac partial diploid analysis:
bacterial O+Z-; plasmid O-Z+?
bacterial O-Z+; plasmid O+Z-?
abnormal expression! thus it’s not enough to have a normal copy of each; they must lie on the same chromosome — cis. phenotypically, b-gal is on all the time
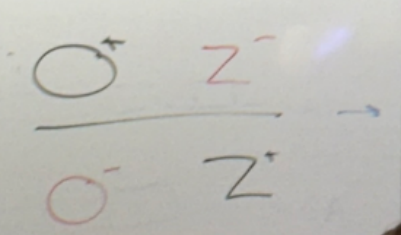
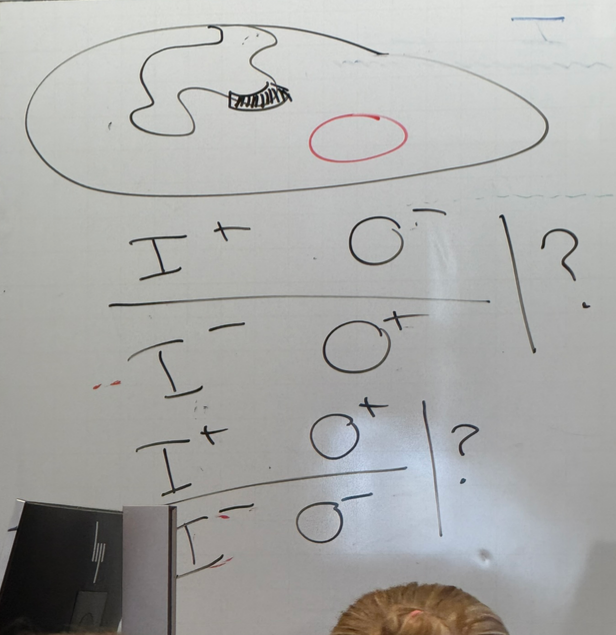
lac partial diploid analysis:
bacterial I+O-; plasmid I-O+?
bacterial I+O+; plasmid I-O-?
bacterial I-O-, plasmid I-O-?
the two strains drawn (first two listed) both behave the same; normal phenotype
thus we need a normal copy of each for normal expression, but don’t have to be on the same chromosome — works in trans! lacI codes for a protein, which can diffuse to any other chromosome.
when no normal copies are present, b-gal is expressed all the time regardless of lactose presence when grown on glycerol
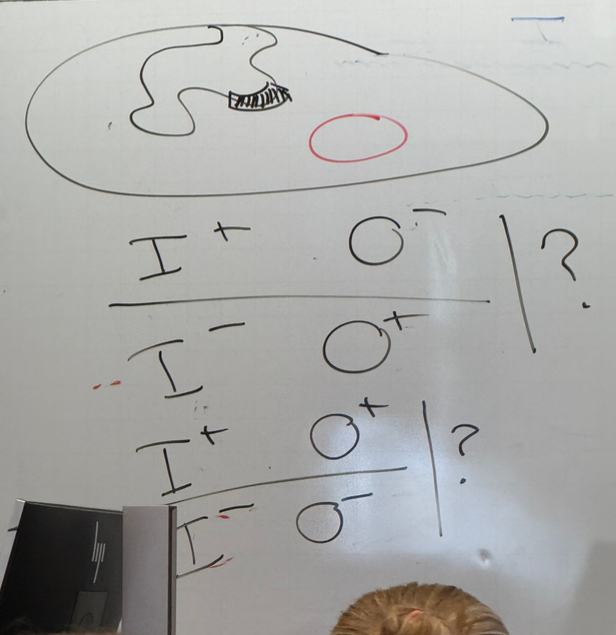
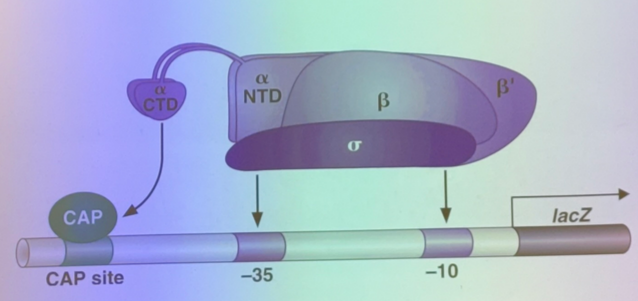
the previous observations for e. coli lac activity change when grown on glucose instead of glycerol — how do we build a genetic circuit that is sensitive to two compounds (lactose and glucose) at the same time?
CAP protein is a TA that stabilizes RNA polymerase binding at promoters. it is glucose-dependent — when glucose is low, cAMP is high. cAMP binds to CAP and allows it to bind to the cis-acting binding site upstream. thus, expression is positively driven.
what does b-gal expression look like under the following conditions?
low lactose, low glucose
low lactose, high glucose
high lactose, low glucose
high lactose, high glucose
low/low: no transcription; lacR is bound and inhibiting RNA pol binding
low/high: no transcription; lacR is bound and inhibiting RNA pol binding
high/low: high expression! lacR is not bound, and CAP is stabilizing the RNA pol binding!
high/high: “leaky” expression — lacR is not bound so RNA pol II can bind, but it’s not stabilized. expression occurs, but at severely reduced rate.
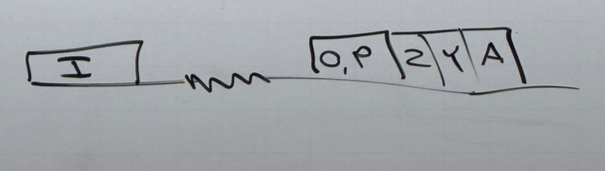
what does each gene in lac code for?
P is the promoter to which RNA pol II binds. O is the operator; the other protein binding sequences
Z encodes b-gal
Y encodes a permease that allows lactose to cross membranes better (transporter protein)
we don’t know what A does
I codes for the lac repressor protein lacR. it has its own promoter. if no lactose is around, lacR protein binds at the operator and blocks RNA pol’s ability to bind to the promoter. if lactose is present, it binds to lacR and changes its conformation such that it can no longer bind the operator, and RNA pol is no longer blocked.
inducible vs repressible systems
inducible: presence of a compound induces expression of the genes necessary to metabolize that compound (i.e. lactose presence drives expression of b-gal, which breaks down lactose)
repressible: presence of a compound blocks synthesis of the same compound (i.e. AA synthesis — once we’ve got enough of a particular amino acid, we don’t need to make any more, so synthesis is shut off)
define combinatorial control and give an example
a combination of signals determine if a gene is expressed or not. for example:
lacZ depends on concentration of lactose and glucose.
TF/morphogen gradients
regulatory map/web/whatever of maternal effect/gap/pair-rule/segment polarity genes
miRNAs found in specific regions of body
transcriptional regulators necessary for cell differentiation (i.e. fibroblasts into myocytes)
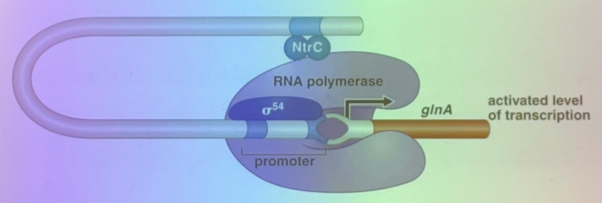
what is this image depicting and how does it relate to gene regulation?
an inducible system involved in nitrogen metabolism. gene expression occurs only if RNA pol can bind. there is a sigma 54 (like sigma 70) that encourages pol binding, and there is a TA several hundred bps downstream… but that distance is okay because they’re far enough apart that DNA can bend to bring the promoter and the TA enhancer together.
how do (some) transcriptional regulators regulate degree to which the helix is teisted?
the MerR protein in a TR that, when bound to mercury, undergoes conformational change that affects the degree to which DNA is twisted. this brings sequences in the major groove into the right orientation to maximize RNA pol binding
true/false: the difference in genetic cell identity between lymphocytes and neurons is due to differences in the genetic code. give evidence
false— both are genetically identical (ignoring antibody junk)
leading theory was that the embryo has all the genes necessary to create any body part, but the cells only retain the genes that are relevant to whatever organ/structure they differentiated to. we know this is wrong bc you can stick a plant in some neglected dirt and it will grow an entirely new plant.
similar stories abt salamanders, twins, and frogs — if you cut off one leg, you can grow a new salamander from that leg. analogous to identical twins (1 egg, 1 sperm, both grow and create two identical “clone” offspring. vanishing twins occur when two embryos fuse — can either create conjoined twins or no visibility at all. thus, some of us in class may be chimeric. frogs: can take an egg, replace the nucleus with one from the skin cells (or any others). will grow to be a regulator tadpole regardless.
do insulators tend to incr or decr gene expression?
tend to squash gene expression when bound by proteins far from the promoter — can be hundreds to hundreds of thousands of bps away!
e. coli vs euks “light-switch” analogy for gene regulation
e. coli and other proks are “near-binary” — either off or on, and can be flipped back and forth
euks have more combinatory control, so it’s more like a rheostat/dimmer switch. can also be tunable to a degree that most prok genes cannot — “turning it up to 11”
what did the scientist Shinya Yamanaka discover about cell differentiation?
lost the race to pull stem cells from human embryos and grow them in a lab to differentiate into the 3 major tissue types, but he was the first to find that you can just make them yourself instead — human induced pleuripotent cells (HIPs)
pulled out an epithelial cell and had to “de-differentiate” it using “4” TAs (or so he believed; we know now that only 3 are necessary). the produced stem cells will grow forever and have induced pleuripotency. on some level, this happens at least partially in all tumors, particularly regarding metastasis. tumor cells must de-differentiate so that they might gain characteristics of different tissues wherever they land. also think of teratomas — tumors that can grow teeth/hair/etc
what is the use of induced pleuripotent stem cells?
can take a cell from yourself and turn it into a stem cell that can grow any tissue type. can use this to study a genetic disorder, or turn them into a cell type that you are lacking and transplant it back into yourself.
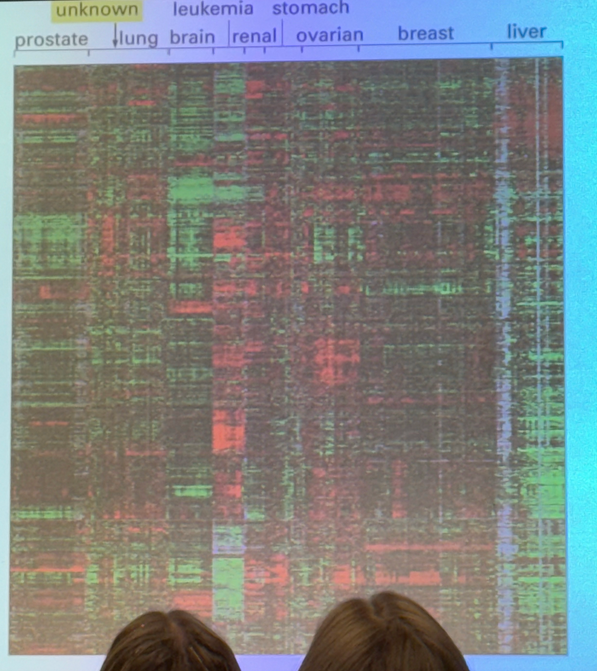
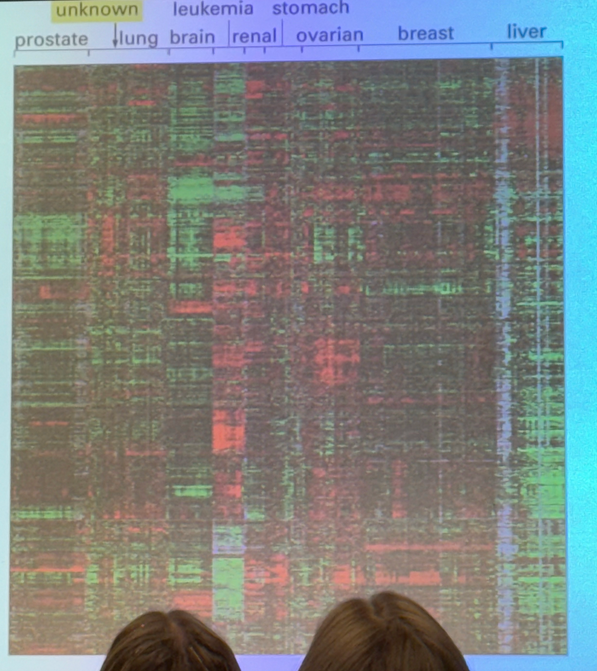
what does a heat map show us?
looks at 10k genes simultaneously. y axis has thin lines stacked, and each corresponds to one gene. moving left to right, we see cells derived from tumors from a variety of different tissues. colors tell us the ratio of how much of a particular gene is expressed in the tumor vs a non-tumor in the same organ. if it expresses more than the normal, we see green. if the tumors express less of a gene than we see in a normal cell, we see red. in between, no color at all. not all identical to each other… but we can compare. i.e. we see a chunk of green genes all together, and we can compare to lung tumors where those same genes are underexpressed (red) vs breast tumors that don’t show much difference in expression compared to the regular cells.
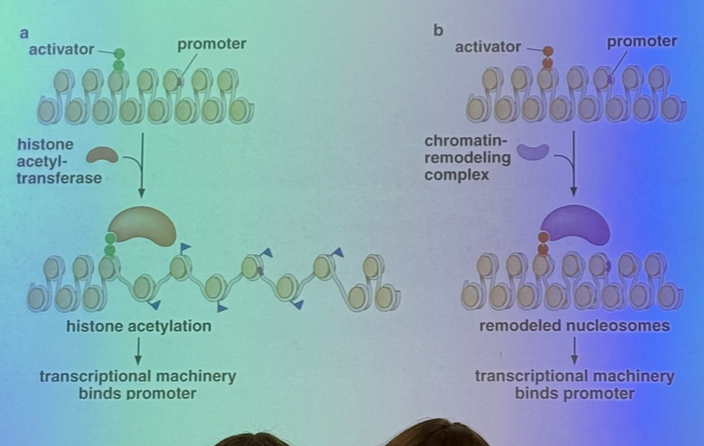
chromatin structure obviously affects expression, but how can TAs change the DNA packing to increase expression in a particular sequence?
a TA can bind to its enhancer and bring in a histone acetyltransferase (HAT) that will add acetyls to histone tails, which generally leads to unpacking and relaxation of the nucleosomal structure!
doesn’t act on just one nucleosome either — the HATs tend to sit on acetylated nucleosomes in order to activate their neighbors
they can also bind to chromatin remodeling enzymes, which act like a pulley/spool to pull DNA off of the nucleosome (CONNECT TO PREVIOUS UNIT!)
(CONNECTION TO PREVIOUS UNIT!)
describe histone modifications + explain how to think of the histone core regarding spooling
phosphorylation — decreases interactions with DNA due to the negative charges repelling each other
methylation — can drive to either euchromatin or heterochromatin, depending.
histone methyltransferases (add methyls) are writers
histone demethylases (remove) are erasers
acetylation — drives to euchromatin; associated with incr expression!
think of histone core as a wheel with a fixed axle. can spin it so that the promoter sequence (for example; it works for any sequence for which a protein binds in a sequence-specific way) spools off of the wheel.
how can transcriptional repressors affect DNA packing to decrease expression of a particular sequence?
calling in histone deacetylases to de-activate the sequence and push it towards heterochromatin
can reverse the actions of the chromatin remodeling complex
a typical human gene might have roughly how many enhancer sites for TAs to bind?
~a dozen enhancers
we can imagine there may be several repressors and/or inhibitors per gene too, but don’t know how many
do all genes require that chromatin structure is changed in order to have increased expression?
no — some genes already have the loose, relaxed structure for transcription
i.e. in HIPs (human induced pleuripotent cells), ALL genes necessary for differentiation into any of the three tissue types are ALL available for expression
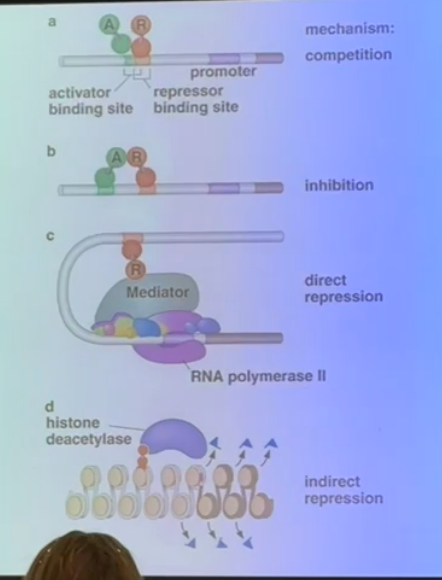
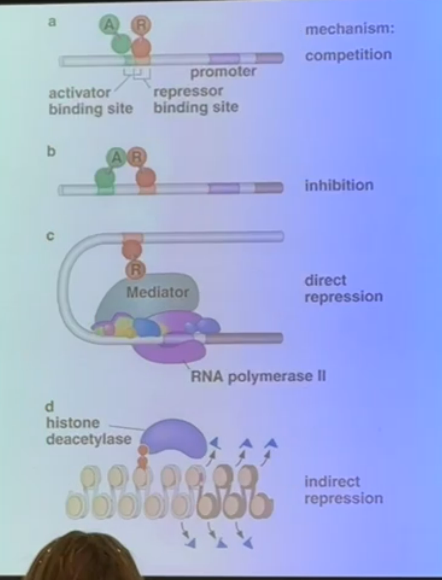
explain the different methods used by transcriptional repressors seen on this slide
a) if a repressor binding site overlaps with an enhancer, the repressor can come in to bind and force off the activator
b) a repressor can bind at a site that is physically separate from the enhancer, but still directly interact with the activator to inhibit its functionality
c) interacts with the mediator complex can cause direct repression by preventing RNA polymerase or any activators from binding
d) removing acetyl groups encourages tighter chromatin structure to squelch gene expression
broadly, what do insulators do?
determine which trans-acting activators can/cannot act on which promoters, even from very very far away
enh have to come in contact w promoters to work. insulators affect that ability
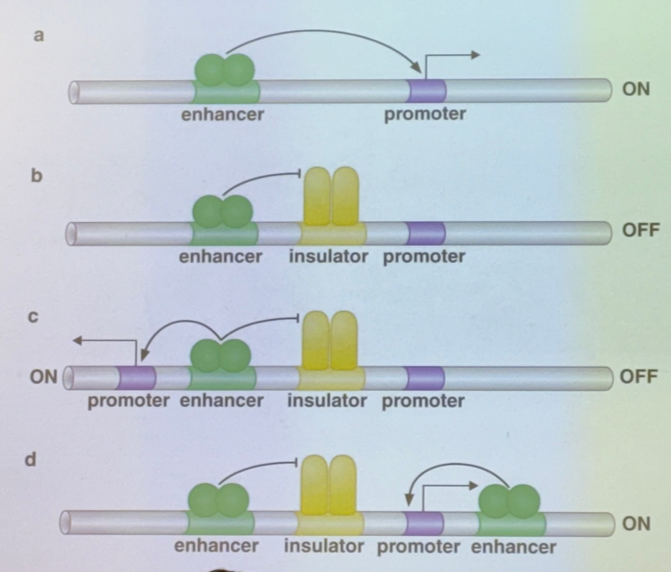
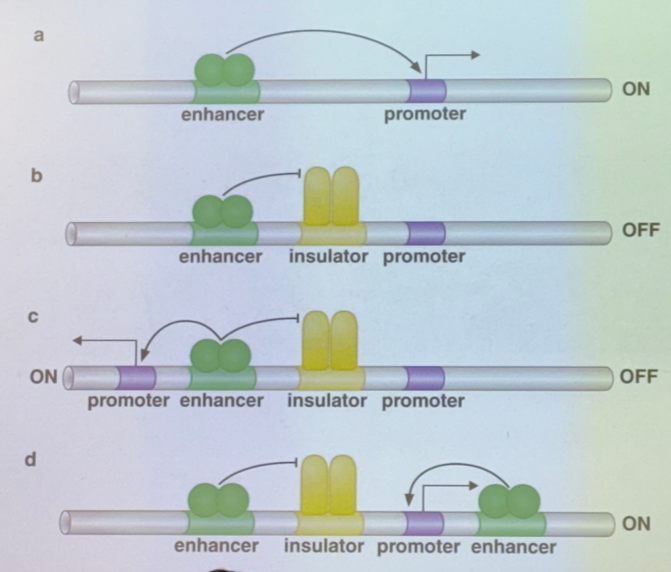
explain each shown method by which insulators can affect gene expression
a) no insulator; TA binds to enhancer to activate the promoter
b) there’s one enh, one insulator, and one promoter. the insulator blocks the activator’s action
c) there’s one enh, one insulator, but two promoters flanking. the insulator blocks one promoter and not the other, so that one enhancer will only activate one or the other (choose one sequence to enhance over another)
d) there’s two enh, one insulator, one promoter. the insulator blocks one enhancer and not the other, so only one TA can act (choose one method of activation over another for the same sequence)
it is VERY IMPORTANT to note that there are ~12 enhancers per human gene, so there are many different combinations for what enhancers/promoters can act or not! particularly relevant for scenario c: even though the insulator can only pick one promoter to be enhanced, there are other enhancers that can come in and turn on the other one too. thus it isn’t truly a case of “only one of the two can be activated at once”
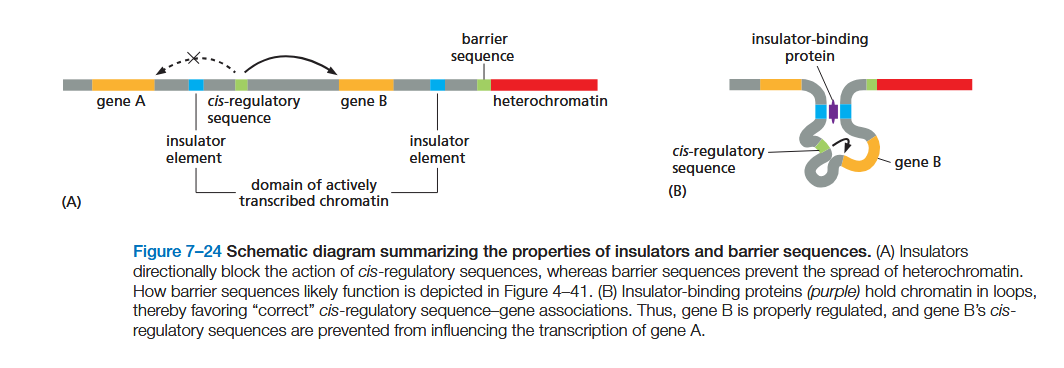
describe in a more 3D way how insulators work
Insulators function by forming loops of chromatin, an effect mediated by specialized proteins that bind them (see Figures 4–48 and 7–24B). The loops hold a gene and its control region in rough proximity and help to prevent the control region from “spilling over” to adjacent genes.
in the yeast research paper, how many promoters does each activator bind?
varies enormously, but broadly speaking, out of 8k total promoters:
~1/3 of the TAs bind 1-20 promoters
~1/3 of the TAs bind 21-40 promoters
~1/3 of the TAs bind 41-180
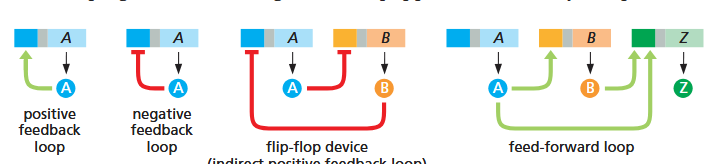
in the yeast research paper, what are 6 of the motifs seen in gene regulation?
autocatalytic loop — regulates expression of itself as a positive feedback loop. good for changing behaviors quickly and irreversible (antihomeostatic). ex: production of yeast “gametes”; anaphylaxis
single-input motifs — one TF, when activated, can regulate a set of multiple genes. (push one button to get many responses). i.e. Leu3 regulates 3 genes
multi-input motifs — multiple genes are regulated each by multiple genes, i.e. 4 genes are regulated by 3 different TAs in a web. two ways for this to work — either we don’t need all three and they’re just there for contingency, OR all three need to be active and thus limits the conditions under which this pathway could occur. both are possible!
multi-component loops — another form of positive feedback loop forming a square/circle. A > b > C > d > A …
feed-forward loop — one gene both directly and indirectly regulates another. A > b > C > d, but also A > d directly
chain — specific order of operations that must occur one after another, one expressed at a time. like making a sandwich. A > b, then b > C, then C > d, then d > E, then E > f. i.e. mitosis, cell cycle as a whole
yeast research paper: what are the 3 groups into which they grouped the ~106 TFs based on known function?
developmental genes — interregulate each other to significant degree, and regulate some others (including a few from metabolism)
metabolism genes — tend to regulate each other extensively. regulate other systems too, but they are very tightly linked to each other
cell cycle genes — regulate each other and organized into the sequential/chain pattern. also regulate TFs from other groups
did not discuss others, though they almost certainly exist
roughly what percent of the human genome is dedicated to regulating itself?
~15%. true for all multicellular organisms too
generally speaking, how is it that in development we reproducibly create an organism with our degree of polarity (head/tail, proximal/distal, ventral/dorsal, etc)?
through gene networks where TFs regulate expression of other TFs which regulate expression of genes to create particular body parts in particular locations
gene expression patterns (bicoid, gap, pair-rule, segment polarity, hox) that divide into finer and finer segments!
define morphogens
hormone-like molecules that regulate production of morphological structures
we know now that morphogens turn on TFs, and are thus close in function to TFs themselves. Turing used them in his model to describe polarity
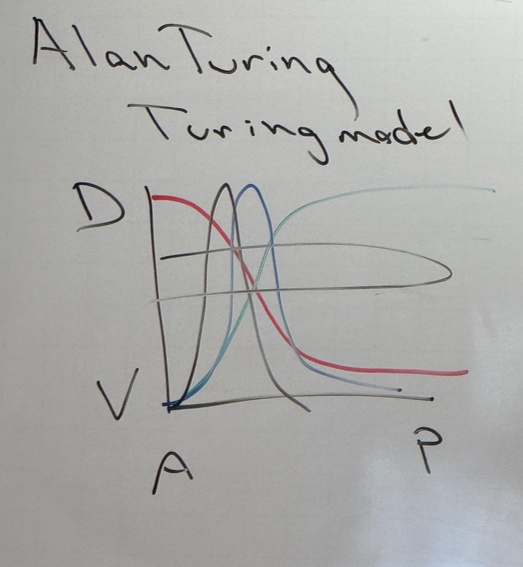
explain the turing model
morphogens/TFs can exist in gradients — one is activated at one end, another at the other end (red and green). because we have combinatorial control, we can have TFs that are dependent on particular concentrations of both morphogens (blue). we could also have a TF that depends on the concentrations of red and blue (shown in black). etc etc etc
the same story can apply across other axes, i.e. P/D, V/D, etc. for each, we would have another set of peaks. thus, we have more and more highly specific location-based gene expression
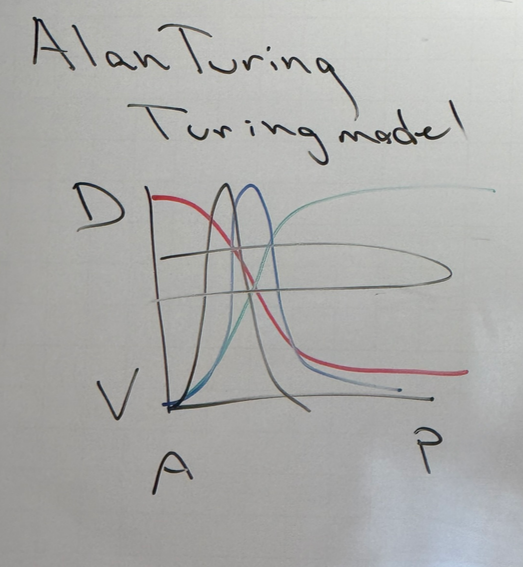
explain how cheetahs get their spots, zebras/tigers get their stripes, etc
have a positively self-regulating TA that also activates a partner that is inhibitory towards the activator. if they diffuse at different rates, we would see a zone of activation where the concentration of the activator is high enough. as you move further and further away though, you have less expression of the TA. have a halo of inactivation around the central zone
this story isn’t so important; meant to be an intro to the stripe patterns in drosophila development.
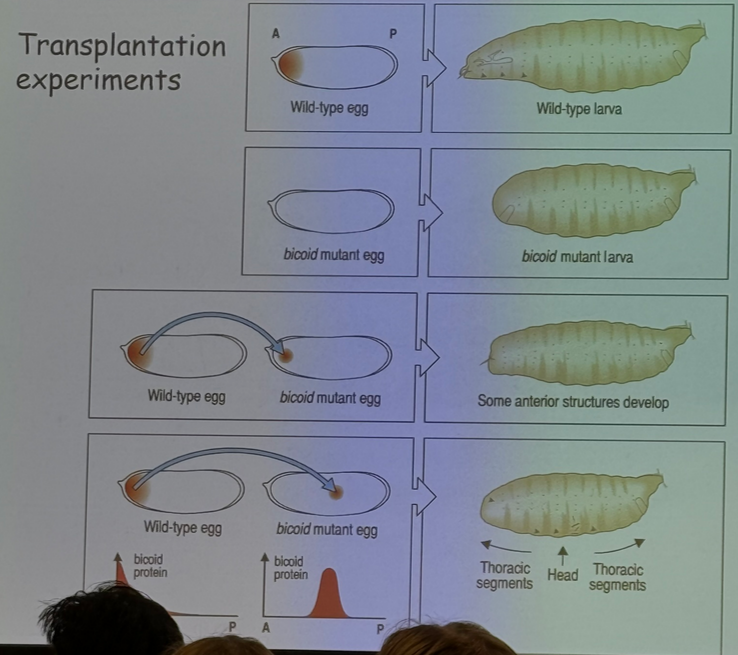
describe the experiment shown
bicoid is associated with anterior/posterior patterning, specifically anterior/head structures
a bicoid mutant larvae does not grow a head. if you transplant some bicoid material from a wild-type head into the anterior of a mutant, some anterior structures start to develop.
if you transplant some bicoid from the wild type head and insert it into the middle of the egg, however, the head starts forming in the middle of the thorax.
additionally, not shown here but if you transplant bicoid into the middle or posterior of a wild type egg, it’ll appear to form two heads (at least for a while)
in complex organisms like drosophila or humans, are eggs polar before fertilization, or are they made polar by the binding point of the sperm during fertilization?
in complex organisms, the egg is already polar — maternal-effect genes
some simple organisms do exhibit the other story though, where the egg is uniform until sperm binds and creates a structure that determines the orientation of the rest of the organism
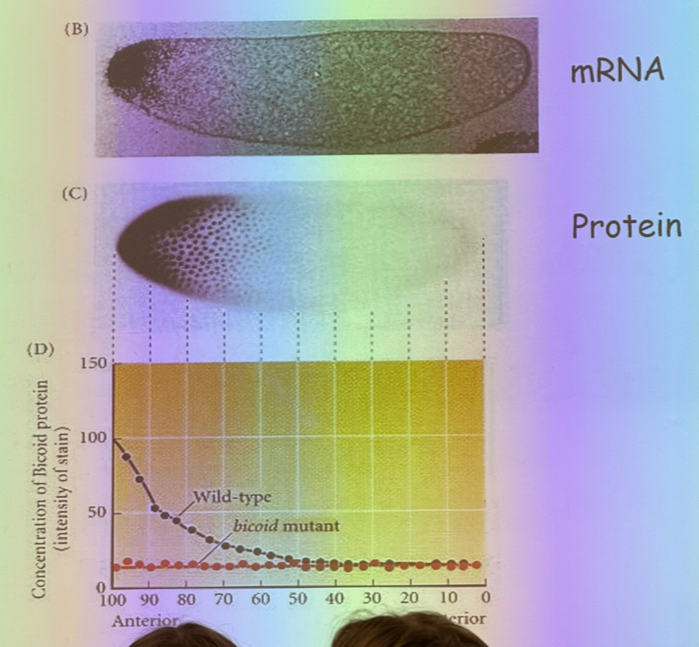
explain this image
mRNA is found at one end of the egg and not the other thanks to motor proteins that drag them off to one side. if mRNA is conc on one side, then the proteins that they encode for are free to diffuse. shortly after fertilization, we have syncitial development where we have mitosis without cytokinesis — 1 cell, 1k nuclei. bicoid is a TF that does its job in the nucleus and is free to diffuse.
this is important because if we increase temp, then diffusion increases and we get more bicoid on the opposite end… but we don’t get flies with bigger heads, so what is missing? — must be another factor working the same way from opposite end. thus its diffusion would also increase, and the center of overlap would be the same
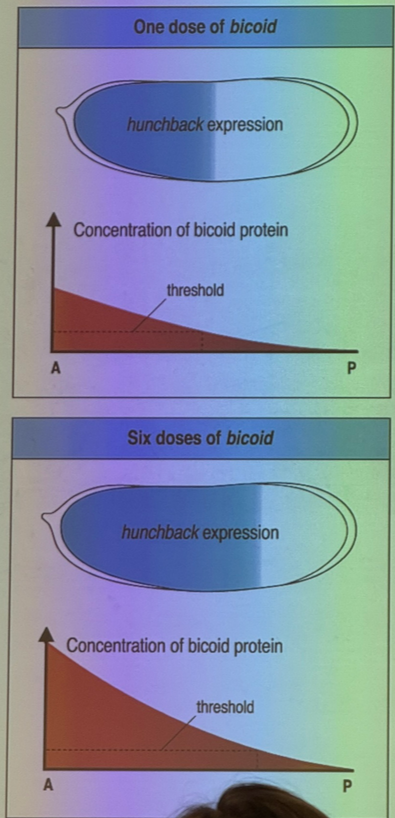
since the effect of increased temperature on bicoid distribution/effect gets cancelled out, what can we do to affect the phenotype?
change the dose of bicoid
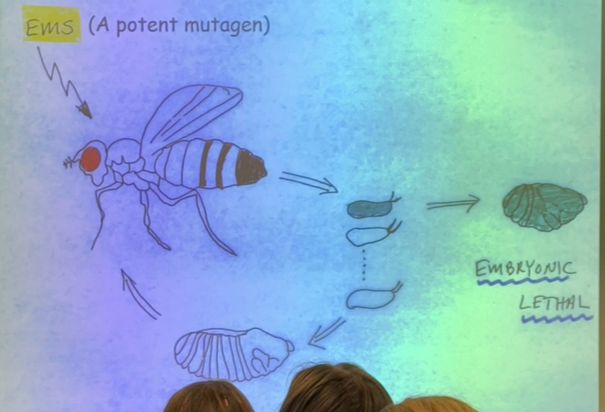
explain this image
randomly mutated mother flies and saw which mutated eggs led to developmental defects, particularly embryonic lethals, which don’t develop at all. categorized into groups: gap gene, pair-rule, and segment-polarity
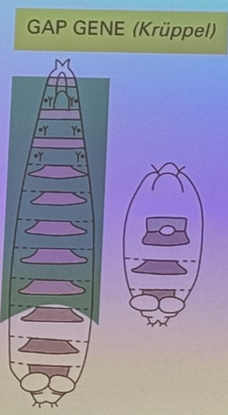
what happens when gap genes are mutated? example?
large segments where the middle of the embryo is gone. i.e. kruppel and hunchback
what happens when pair-rule genes are mutated? example?
deletion of every-other body segment. i.e. even skipped, where segments 2, 4, 6, 8, etc are missing. other examples are odd-skipped and fushi terazu
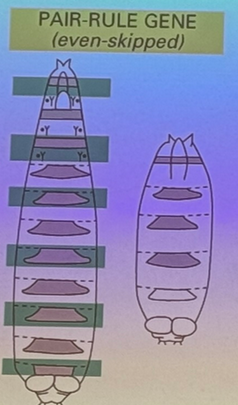
what happens when segment-polarity genes are mutated? example?
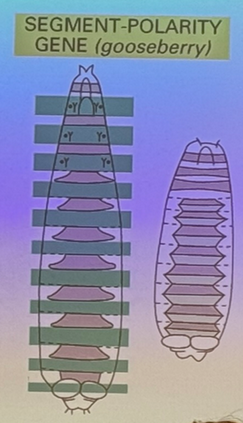
each pair-rule segment is itself polar (anterior/posterior side to each). in this mutation, one half of each segment is affected (i.e. the posterior end of each segment is missing). i.e. gooseberry
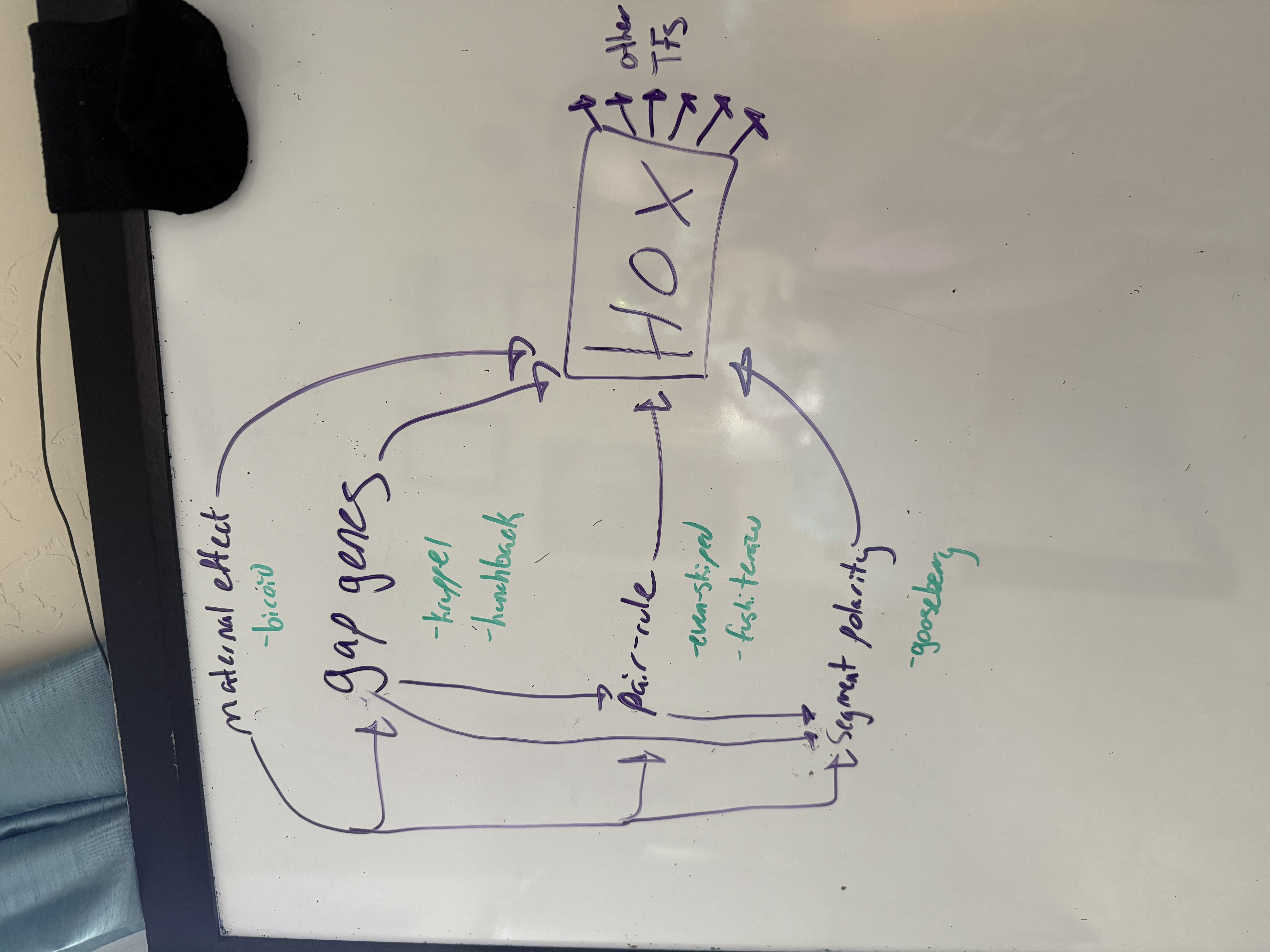
how are the different segmentation genes interconnected? (diagram) — NOT showing activation/inhibition, just regulation in general!
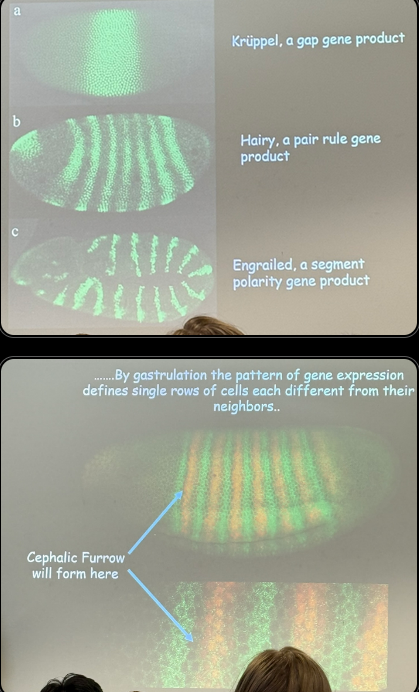
describe this image, including roughly how wide each segment is
gap gene — wide chunk, but pretty specific (can have high expression in one, while its neighbor’s is low). band is 10s to 100s of nuclei wide
pair rule — kruppel is still active, but not seen in some, hence how we can get stripes that are different from each other (one can be hairy + kruppel, one can be hairy and not kruppel). 10s of nuclei wide or less
segment polarity — even more specific and even more potential combos of active vs inactive expression in each fluroescent segment. each band is ~2-3 nuclei wide
eventually we get a pattern that defines single rows of cells differently from any neighbors; each layer is essentially 1 nucleus thick. the cephalic furrow shows the exact location where the head develops (there and ONLY THERE, absolutely nowhere else, no wiggle room for a neighboring stripe).
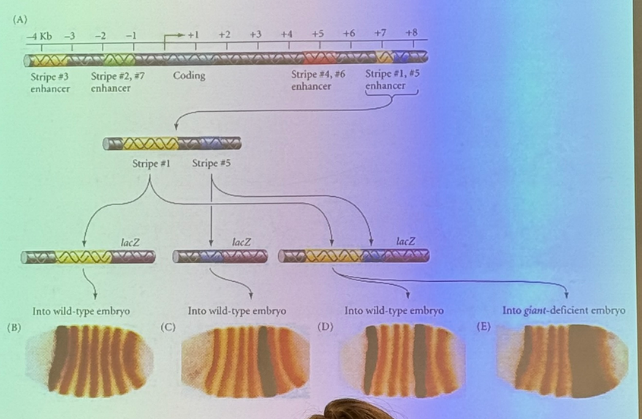
in this diagram for the structure of eve in drosophila, the metallic blue sequence is the actual gene itself (coding seq). there are several enhancers upstream and downstream which are necessary for driving expression of the gene. we get rid of each one and see which stripes, if any, go away
it’s easy to say one enh is necessary for expression of stripe 3 (for example), but maybe it works collaboratively with the other enhancers at the other stripes. how can we tease that out?
replace it; if it works it is alone sufficient enough to generate that behavior/structure — entire machinery necessary is there bc you have added nothing else.
if you replace it and can’t put it back the same way without affecting other structures, you know the overlap =combinatorial control for those other structures
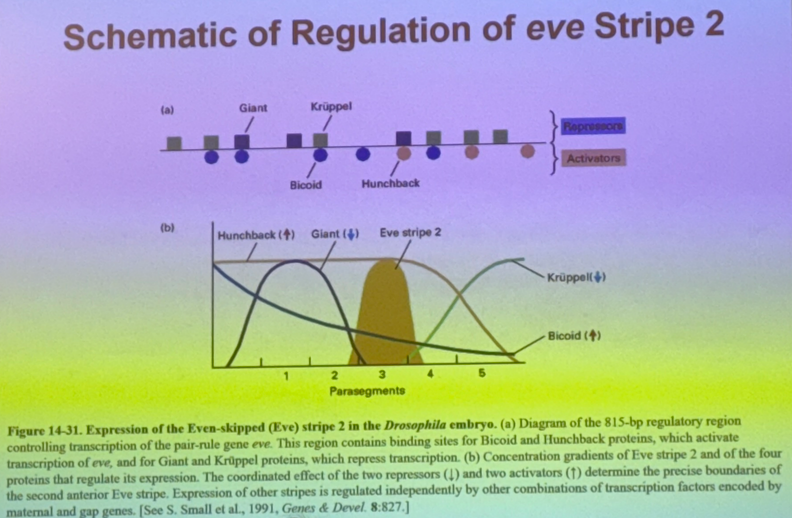
in order to express the gene for eve stripe 2 in a particular location, then we need a mechanism to drive expression here and inhibit expression elsewhere in order to have fine order of control. how is this accomplished?
a maternal gene regulates the expression of 3 gap genes which regulate expression of even-skipped. this particular order allows its expression in the stripe, and a different order will NOT allow expression elsewhere.
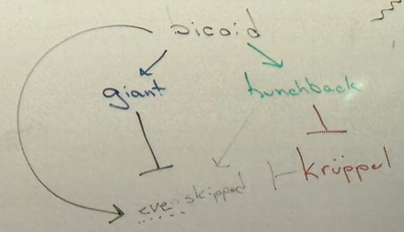
draw the activation/inhibition interactions relating bicoid, giant, hunchback, even-skipped, and kruppel
be able to draw this from scratch without notes — should allow you to answer most questions in-depth!
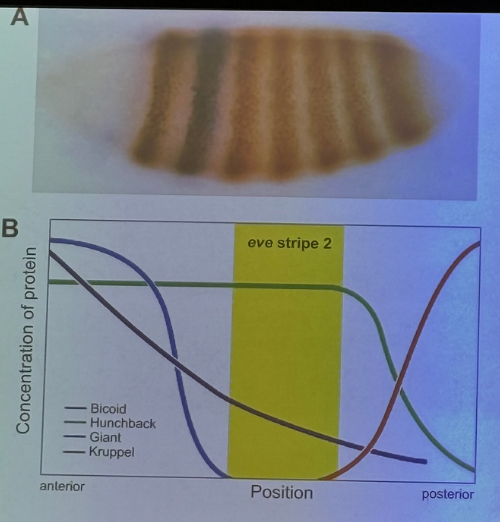
draw the diagram showing the expression of bicoid, giant, hunchback, even-skipped, and kruppel along the A-P axis, then mark where even-skipped stripe 2 appears
insert
**draw this yourself so the lines/colors can label better. reference from 4/28
be able to draw this from scratch without notes — should allow you to answer most questions in-depth!
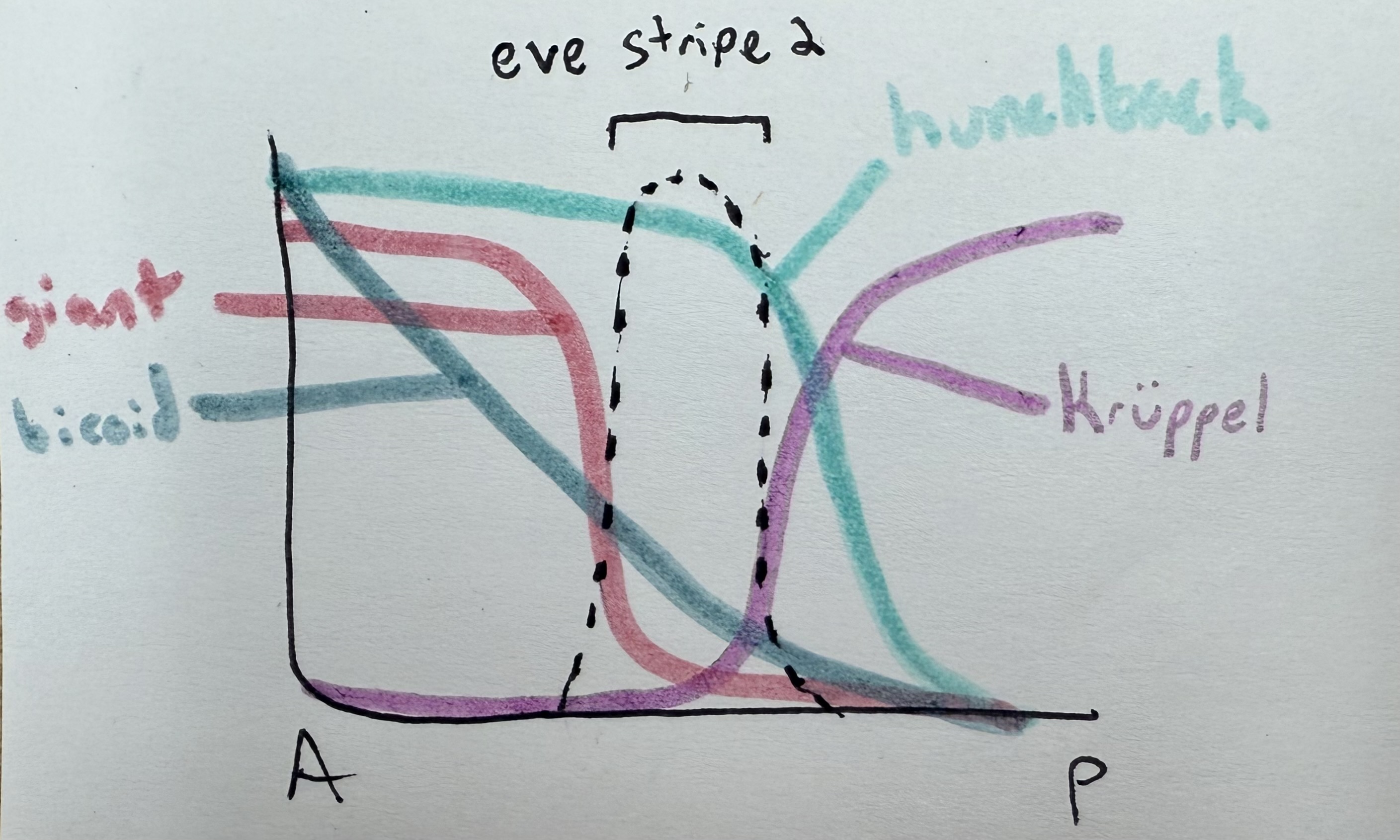
how does bicoid affect hunchback vs giant?
bicoid turns on hunchback. very strong thresholding effect. get expression of hunchback only when we have enough bicoid — threshold is relatively low though, don’t need a ton of bicoid to drive hunchback.
bicoid similarly turns on giant. thresholding effect, but threshold is much higher.
how does hunchback affect kruppel vs even-skipped?
inhibits kruppel
activates even-skipped
think about how a protein can be both an activator and an inhibitor, depending on the context it’s in
some proteins (i.e. the CAP protein) can act as both a repressor and an activator, depending on the exact placement of their enhancer sequence relative to the promoter: for some genes, the CAP enh sequence overlaps the promoter, and CAP binding thereby prevents the assembly of RNA polymerase at the promoter.
how is even-skipped affected by the other four in the pathway (bicoid, giant, hunchback, kruppel)? thus, where will we see even-skipped?
bicoid activates
giant inhibits
hunchback activates
kruppel inhibits
→ we see even skipped where there is bicoid and hunchback, and no giant or kruppel
more than one copy of each protein binds at the enhancer, and the repressors that bind will bind also to the DNA.
explain 3 of the methods by which the repressors can work — 1 less likely, 2 more likely
can repress even-skipped to avoid expression of stripe two by binding to DNA and keep RNA pol II from binding (but probably not this).
could bind and modify histones and convert to heterochromatin.
could bind at sites that other transcriptional activators would normally bind to.
many more possibilities. this is the inherent challenge in understanding gene expression in euks, esp complicated euks.
classical examples of epigenetics
crocodilian sex ratio is determined by temperature at which eggs are incubated
clownfish gender is plastic
what is the relative permanence of epigenetic effects?
some permanence — get passed down minimally from cell to cell, and maximally from individual to individual
true/false: there is a lot of cellular memory for TF changes
false. we can change the activity of a TF, and we can usually reverse it… but even if we can’t reverse it, it’ll probably die soon anyway
true/false: there is a lot of cellular memory for changes in chromatin structure
some TF regulate enzymes that control chromatin form, and the chromatin structure is passed down from cell to cell — highly conserved!
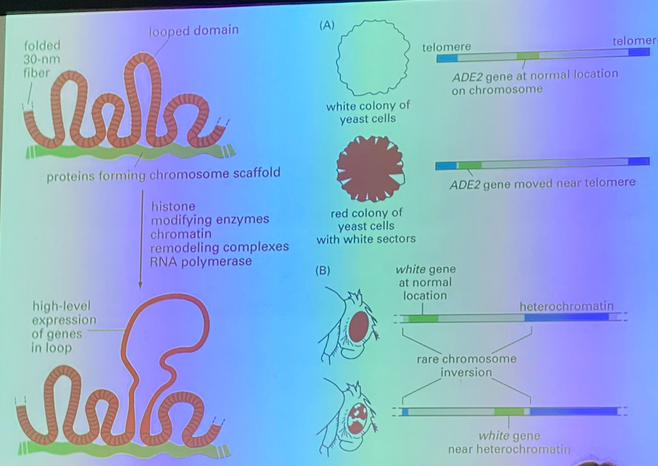
connect to previous units — the red/white yeast experiment!
when DNA is replicated, the histones are displaced. half of the histone proteins are left behind and new proteins have to come and bind. the ones left behind bear modifications from the previous cell. those modifications remain and provide signals to recapitulate machinery to recreate the modifications seen before the S phase.
if we can take the cause of some environmental insult and activate a TA that converts heterochromatin to euchromatin, we can do the opposite. only need to activate it once. can be passed from cell to cell.
(CONNECTION TO PREVIOUS UNIT!)
example of cellular memory regarding chromatin structure
red-white yeast experiment
ADE2 expression = white cells. move this gene seq near telomere (heterochromatin) = no expression = red cells
eventually, white cells appear and have white offspring. thus the chromatin structure has changed enough the ADE2 is slightly moved away from the heterochromatin region and can be expressed again, and it is highly HERITABLE!
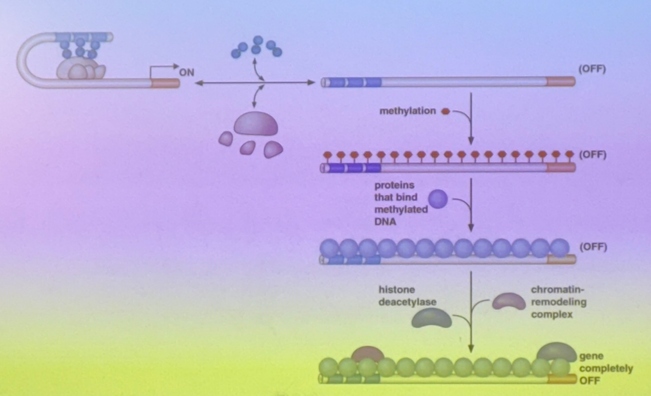
review previous units and DNA methylation, particularly CG/GC dinucleotides
the Cs get methylated, and then if they get deaminated, they turn into T and are difficult to repair. so why do we methylate them at all?
the Cs that are methylated regulate protein binding
imagine that there are lots of CpG islands in the enhancer (dozens of CG dinucleotides). if one gets methylated, almost all of them in the region will be. C has to be able to continue to bind to G though, so the methyltransferase adds it off to the side in the major groove.
thus when Cs are methylated, proteins bind more poorly → the TA is less likely to bind to the methylated enhancer, so even if the TA is turned on, it doesn’t matter if it can’t bind. the gene is repressed even as we take away the methylating signal because the DNA has memory and all copies will look like its predecessor
**ADD REVIEW ON CG/GC DINUCLEOTIDES, CpG ISLANDS, ETC
in addition to blocking TAs from binding to enhancers, how does DNA methylation (particularly cytosines) repress gene expression?
histone deacetylases tend to bind to methylated C. they then remove acetyls from histones, pushing towards heterochromatin